BASES MOLECULARES DE LA DEPLECIÓN DE
GLUTATIÓN Y DE LA NECROSIS CELULAR EN LA
PANCREATITIS AGUDA. PAPEL DE LAS PROTEÍN
QUINASAS ACTIVADAS POR MITÓGENOS
JAVIER PEREDA CERVERA
UNIVERSITAT DE VALENCIA
Servei de Publicacions
- D. Jose Viña Ribes
- D. Daniel Closa Autet
- D. Antonio Manso Martin
- D. Jesus Osada García
- D. Jose Luis Rodríguez García
Va ser dirigida per:
D. Juan Sastre Belloch
D. Gerardo López-Rodas
D. Luis Sabater Ortí
©Copyright: Servei de Publicacions
Javier Pereda Cervera
Depòsit legal:
I.S.B.N.:978-84-370-6682-0
Edita: Universitat de València
Servei de Publicacions
C/ Artes Gráficas, 13 bajo
46010 València
Spain
DEPARTAMENTO DE FISIOLOGÍA
Bases moleculares de la depleción de glutatión y de la
necrosis celular en la pancreatitis aguda. Papel de las
proteín quinasas activadas por mitógenos.
JAVIER PEREDA CERVERA
TESIS DOCTORAL presentada para la obtención del
grado de Doctor en Farmacia
AGRADECIMIENTOS
Han sido muchos años de trabajo para la realización de este trabajo y muchas las personas que han contribuido en él. Por eso, quiero expresar mi más profundo agradecimiento a todos aquellos que han hecho posible que llegue este momento.
Quiero agradecer especialmente a mis directores de tesis, Juan, Gerardo y Luis:
A Juan, porque has sido la guía y el alma de esta tesis. Has sido la luz cuando veía las cosas oscuras y me has enseñado a ser científico de la mejor manera. Cada año que ha pasado me ha mostrado tu calidad como persona, tu espíritu de trabajo y tu entusiasmo. Muchas gracias por todo, Juan. Tienes mi más profunda admiración y respeto.
A Gerardo, por tu gran implicación y tus sabios consejos. Por tu paciencia y por abrirme al mundo de la genética de forma tan fascinante. Me he sentido un “cromatina” más en tu laboratorio siempre con tu ayuda. Muchas gracias, Gerardo.
A Luis, porque le das sentido a tanta molécula y a tanto dato. Me has enseñado a sacar la esencia de las cosas y buscar lo importante de la forma más sencilla. El servicio de Cirugía General del Hospital Clínico ha sido una segunda casa para mí. Muchas gracias, Luis.
A Pepe, por acogerme en tu grupo y por enseñarme a pensar como un científico. Echo de menos las reuniones de los viernes, a veces tan terroríficas y a veces tan geniales.
A Fede, por ser tan buena persona, por darle humor a la vida y a los problemas. Has dejado en mí una gran huella. Lástima que no seas un “pancreático”.
Víctor y tú habéis tratado esta tesis como si fuera vuestra. A Laia, por ayudarme tantas horas y por estar cerca a pesar de estar tan lejos.
Como no, quiero agradecer al resto del laboratorio que tanto han contribuido en esas pequeñas cosas que son tan grandes. Gracias, M. Carmen, has sido un referente para mí en trabajo y dedicación. Gracias por las charlas que hemos tenido juntos. A Ana, que me ha enseñado tantas cosas de la vida y de la ciencia. A Chelo, por su sacrificio continuo. A Diana por su lealtad incondicional, su entusiasmo y su arrojo por la ciencia. Tienes mi admiración y mi amistad. A Soraya por su manera de ver las cosas. A Juan Gambini, Marco y Alfonso por su amistad y por dar un poco de testosterona entre tanto estrógeno. A Jelena, a la que admiro por su valentía y a Nancy que recuerdo con “la rubia tarada”.
Me gustaría hacer un agradecimiento especial al grupo de José Estrela. Muchas gracias, Ángel por los experimentos de confocal, pero sobre todo por tu amistad y compañerismo durante tantos años. A Julián, modelo a seguir, gracias por enseñarme como científico y como persona. ¡Te echo mucho de menos! A María y Salva que contagian con sus ganas de trabajar.
A todos los que pasasteis por el laboratorio. A Gaetano, que diste un toque tan pintoresco al laboratorio. A Teresa, que tanto ha trabajado conmigo codo con codo y tantas horas hemos pasado hablando de la vida. A Natalia y Belén por su ayuda, os deseo mucha suerte. A Rafa por su ayuda con la xantina oxidasa. También quiero recordar a Puri, Juamba y José Antonio. Empecé cuando vosotros acababais y me mostrasteis un mundo por descubrir.
Un beso muy especial para Juana que tanto ayudó al principio de este trabajo. También muchos agradecimientos a las “secres” y en especial para Elena, que tanto nos ha ayudado con las cosas de palacio y nunca han ido despacio. Otro beso para Inma, que alegra tanto las mañanas. Gracias a todas las de la U.C.I. y en especial a Asun, Consuelo, Inma y Luisa. Siempre han ayudado desinteresadamente.
Quiero agradecer a Antonio Alberola su apoyo y ayuda. También quiero recordar a los dos “Luises”, a Isabel y Paqui. Además de trabajar juntos también hemos disfrutado muchas celebraciones juntos.
Gracias a Juan y Concha, y a todos los de Bioquímica, que siempre habéis estado dispuestos a ayudar.
Gracias al grupo de Pandol en Los Ángeles, y muy en especial a Aure, que tan bien me ha tratado.
No quiero dejar de dar las gracias a toda la gente de Barcelona. En especial muchas gracias a Dani Closa, al que admiro como científico y al que agradezco la oportunidad de hacer una nueva vida en la gran ciudad. Muchas gracias a Ana, que ha sufrido los retrasos de la tesis y de la que he aprendido mucho. También agradecer a Antonio Xaubet, María Molina y Oriol Bulbena por permitirme formar parte de su grupo. Un beso muy grande a todos los del “402”.
Muchos amigos he encontrado en el IIBB, y mando un beso especial a Paty, Nacho, María, Gemma, Montse y Emma y a los de neuro, Noe, Judith y Xavi.
En especial, vull agraïr a Llorenç, per aquest any que hem viscut junts. M’has obert les teues portes en Barcelona i en Castalla. He apres a ser millor persona amb tu, m’has ensenyat molt de la vida. Et trovaré a faltar molt.
A mis amigos de siempre, Gabi, David, Jorge, Jaime, Pablo, Alex, Carlitos, Iván y Parre, por no dedicarles tanto tiempo como me gustaría. Tenemos mucho que celebrar.
A mi familia, que siempre me apoya. No somos cariñosos pero nos queremos mucho. Aquí en Barcelona os echo mucho de menos. Papá y Mama, gracias por permitirme estudiar, y ayudarme a acabar la tesis. Un abrazo muy grande, hermanitos.
INDICE DE FIGURAS ... IX INDICE DE TABLAS ... XV ABREVIATURAS ... XVII
I- INTRODUCCIÓN ... 1
1. PANCREATITIS AGUDA. ... 3
1.1. Definición. ... 3
1.2. Epidemiología y etiología... 6
1.3. Patogenia. ... 7
1.4. Fisiopatología. Efectos sistémicos de la pancreatitis aguda. ... 11
1.4.1. Síndrome de respuesta inflamatoria sistémica (SRIS)... 13
1.4.2. Afectación pulmonar en la pancreatitis aguda... 19
2. CITOQUINAS. ... 21
2.1. Concepto. ... 21
2.1.1. Citoquinas proinflamatorias y pancreatitis aguda... 22
2.1.2. Citoquinas antiinflamatorias y pancreatitis aguda. ... 24
2.2. Factor de necrosis tumoral α (TNF-α)... 25
2.2.1. Estructura y función del TNF-α... 25
2.2.2. TNF-α y pancreatitis aguda... 28
3. RADICALES LIBRES... 31
3.1. Concepto de radical libre ... 31
3.2. Clases de Radicales libres... 31
3.3. Génesis de las especies reactivas de oxígeno... 32
3.3.1. Fuentes endógenas ... 32
3.3.1.1. La cadena de transporte electrónico mitocondrial... 32
3.3.1.2. Reacción de Fenton-Haber-Weiss... 33
3.3.1.3. La xantina óxidoreductasa como fuente de radicales libres... 34
3.3.1.3.1. Estructura enzimática... 34
3.3.1.3.2. Conversión de xantina deshidrogenasa a xantina oxidasa... 35
3.3.1.3.4. Distribución orgánica, celular y transporte de la
xantina oxidasa. ... 38
3.3.1.3.5. Regulación de la actividad enzimática... 39
3.3.1.3.6. Xantina oxidasa y pancreatitis aguda. ... 40
3.3.1.4. Fagocitos activados. ... 42
3.3.1.5. Otros. ... 42
3.3.2. Fuentes exógenas. ... 43
4. ESTRÉS OXIDATIVO... 44
4.1. Concepto de estrés oxidativo... 44
4.2. Estrés oxidativo y daño a biomoléculas. Índices de estrés oxidativo. ... 44
4.2.1. Daño oxidativo a lípidos, proteínas, glúcidos y DNA... 44
4.2.2. Cociente GSSG/GSH como índice de estrés oxidativo... 46
5. ANTIOXIDANTES ... 47
5.1. Antioxidantes enzimáticos. ... 47
5.1.1. Superóxido dismutasas. ... 47
5.1.2. Glutatión peroxidasas. ... 48
5.1.3. Catalasa... 49
5.2. Antioxidantes no enzimáticos ... 50
5.2.1. El glutatión ... 50
5.2.1.1. Estructura química del glutatión... 51
5.2.1.2. Propiedades físico-químicas del glutatión ... 52
5.2.1.3. Síntesis del glutatión y su regulación. ... 53
5.2.1.3.1. γ−glutamil cisteína sintetasa... 54
5.2.1.4. Degradación del glutatión, ciclo del γ-glutamilo... 55
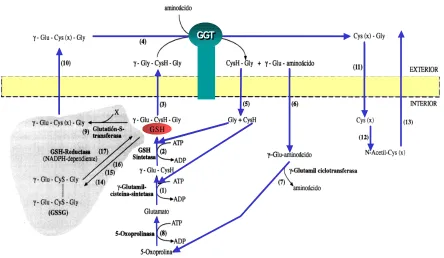
5.2.1.5. Precursores del glutatión ... 57
5.2.1.6. Transporte de glutatión ... 58
5.2.1.7. Localización del GSH... 60
5.2.1.8. Funciones fisiológicas del GSH ... 62
5.2.1.9. Papel antioxidante del glutatión: ciclo redox... 63
5.2.1.10. Glutatión y pancreatitis ... 64
5.2.2. Otros antioxidantes no enzimáticos... 66
6. PROTEIN-QUINASAS ACTIVADAS POR MITÓGENOS (MAP QUINASAS)... 71
6.1. Introducción. ... 71
6.2. Señales de transducción y control de la expresión génica... 71
6.2.1. Rutas de señalización intracelular... 72
6.2.2. Control de la expresión génica. ... 72
6.2.2.1. Factor Nuclear kappa B (NF-κB) ... 74
6.2.2.2. SP-1. ... 74
6.2.2.3. c-Myc... 74
6.2.2.4. c-Jun. ... 75
6.3. MAP quinasas ... 75
6.3.1. ERK 1/2 ... 75
6.3.2. JNK ... 78
6.3.3. p 38 ... 79
6.4. Interacciones entre MAP quinasas ... 81
6.5. MAP quinasas y pancreatitis aguda... 82
6.5.1. Activación de las MAP quinasas en células acinares. ... 83
6.5.2. MAP quinasas en la pancreatitis aguda experimental. ... 85
7. MUERTE CELULAR. ... 85
7.1. Necrosis. ... 88
7.1.1. Mediadores extracelulares, y necrosis. ... 89
7.1.2. Iones en la necrosis... 90
7.1.3. Radicales libres en la necrosis. ... 90
7.1.4. MAP quinasas y necrosis. ... 90
7.1.5. Mitocondria, Bcl-2 y necrosis... 91
7.1.6. Proteínas de choque térmico, proteasas, nucleasas y fosfolipasas en la necrosis. ... 92
7.2. Apoptosis. ... 92
7.2.1. Caspasas y apoptosis... 93
7.2.2. Mitocondria y apoptosis. ... 94
7.3. Apoptosis y Necrosis en la pancreatitis aguda. ... 95
II- OBJETIVOS ... 97
1. MATERIALES. ... 103
1.1. Animales de experimentación... 103
1.2. Aparatos. ... 103
1.3. Reactivos. ... 106
2. MÉTODOS. ... 107
2.1. Modelo experimental de pancreatitis aguda necrótica. ... 107
2.1.1. Descripción del modelo. ... 107
2.1.2. Diseño del estudio de la pancreatitis aguda necrótica. ... 109
2.1.2.1. Grupos de estudio para conocer la cinética de la depleción de glutatión, de la regulación de la expresión génica de la γ -glutamil cisteína sintetasa y de la activación de las MAP quinasas en la pancreatitis aguda experimental necrótica... 109
2.1.2.2. Grupos de estudio para conocer el efecto del tratamiento con oxipurinol y pentoxifilina sobre la activación de las MAP quinasas... 110
2.2. Modelo experimental de pancreatitis aguda inducida por ceruleína. ... 111
2.2.1. Descripción del modelo. ... 111
2.2.2. Diseño del estudio de la pancreatitis aguda inducida con ceruleína... 112
2.2.2.1. Grupos de estudio con ratas para conocer la regulación génica y expresión de la GCS. ... 112
2.2.2.2. Grupos de estudio con ratones deficientes en el del TNF-α o en el receptor 2 del TNF-α... 112
2.2.2.3. Grupos de estudio con ratones deficientes en TNF-α... 113
2.3. Aislamiento y cultivo de células acinares de páncreas de rata. ... 113
2.3.1. Fundamento... 113
2.3.2. Reactivos. ... 114
2.3.2.1. Tampón de digestión: ... 114
2.3.2.2. Medio de cultivo: ... 115
2.3.3. Procedimiento... 115
2.3.4. Diseño del estudio con células acinares de páncreas de rata. ... 116
2.4. Cultivos celulares de células AR42J... 117
2.4.2. Condiciones de cultivo... 117
2.4.3. Procedimiento... 118
2.4.4. Diseño del estudio con células AR42J. ... 119
2.4.4.1. Incubaciones para conocer el efecto del taurocolato sobre el nivel de glutatión en células AR42J. ... 119
2.4.4.2. Incubaciones para conocer el efecto de la inhibición de las MAP quinasas sobre el nivel de glutatión tras la incubación con taurocolato en células AR42J. ... 119
2.4.4.3. Incubaciones para conocer los efectos de la inhibición de las proteasas sobre el nivel de glutatión tras la incubación con taurocolato en células AR42J. ... 119
2.4.4.4. Incubaciones para conocer el papel del cálcio, del proteasoma y de la GGT en la depleción de glutatión tras la incubación con taurocolato en células AR42J. ... 120
2.5. Determinación de proteínas... 120
2.5.1. Fundamento... 120
2.5.2. Procedimiento... 120
2.5.3. Cálculos. ... 121
2.6. Determinación de glutatión reducido. ... 121
2.6.1. Fundamento... 121
2.6.2. Procedimiento... 121
2.6.3. Cálculo. ... 122
2.7. Determinación de la actividad lactato deshidrogenasa. ... 122
2.7.1. Fundamento... 122
2.7.2. Procedimiento... 123
2.7.3. Cálculos. ... 123
2.8. Western Blot. ... 123
2.8.1. Fundamento... 123
2.8.2. Condiciones del gel y transferencia... 124
2.8.3. Visualización... 125
2.8.4. Cuantificación de los resultados... 127
2.10. Retrotranscripción-Amplificación del RNA (RT-PCR)... 127
2.10.1. Fundamento... 127
2.10.2. Retrotranscripción del RNA total. ... 127
2.10.3. Análisis del RNA total retrotranscrito por PCR cuantitativo... 127
2.11. Inmunoprecipitación de fragmentos de cromatina (ChIP). ... 128
2.11.1. Fundamento... 128
2.11.2. Procedimiento... 128
2.11.2.1. Preparación de núcleos en tejido pancreático de rata. ... 129
2.11.2.2. Fragmentación de la cromatina entrecruzada. ... 129
2.11.2.3. Cuantificación del DNA. ... 130
2.11.2.4. Inmunoprecipitación de la cromatina. ... 131
2.11.2.5. Análisis por PCR del DNA inmunoprecipitado... 132
2.12. RNApol-ChIP... 133
2.13. Estudios mediante microscopía confocal. ... 134
2.13.1. Fundamento... 134
2.13.2. Procedimiento y diseño del estudio... 136
2.13.2.1. Estudio de los niveles de glutatión en acinos pancreáticos. .... 136
2.13.2.2. Estudio de los niveles de glutatión y del tipo de mortalidad celular en células AR42J... 136
2.14. Análisis estadístico. ... 137
IV- RESULTADOS ... 139
1. EVOLUCIÓN DE LOS NIVELES DE GLUTATIÓN REDUCIDO EN PÁNCREAS EN EL CURSO DE LA PANCREATITIS AGUDA NECRÓTICA.... 141
2. ESTUDIO DEL PAPEL DE LA γ-GLUTAMIL CISTEÍNA SINTETASA EN LA PANCREATITIS AGUDA EXPERIMENTAL NECRÓTICA.... 142
2.1. Expresión de la γ-glutamil cisteína sintetasa en la pancreatitis aguda necrótica. ... 142
2.2. Unión de la ARN polimerasa a la región codificante del gen de la γ-glutamil cisteína sintetasa en cabeza del páncreas en la pancreatitis aguda necrótica. ... 145
2.3. Unión de factores de transcripción a los promotores de la γ-glutamil cisteína sintetasa en la pancreatitis aguda necrótica. ... 146
3. NIVELES DE GLUTATIÓN REDUCIDO EN PÁNCREAS EN LA PANCREATITIS AGUDA EDEMATOSA EN RATAS.... 149
4. ESTUDIO DEL PAPEL DE LA γ-GLUTAMIL CISTEÍNA SINTETASA EN LA PANCREATITIS AGUDA EXPERIMENTAL EDEMATOSA.... 150
4.1. Expresión de la γ-glutamil cisteína sintetasa en la pancreatitis aguda
edematosa. ... 150 4.3. Unión de factores de transcripción al promotor de la subunidad catalítica de
la γ-glutamil cisteína sintetasa... 151
5. FOSFORILACIÓN DE LAS MAP QUINASAS EN LA PANCREATITIS AGUDA EXPERIMENTAL NECRÓTICA.... 152
5.1. Evolución de la fosforilación de ERK en páncreas durante la PA necrótica. .... 152 5.2. Evolución de la fosforilación de JNK en páncreas durante la PA necrótica... 154 5.3. Evolución de la fosforilación de p38 en páncreas durante la PA necrótica... 156
6. EFECTO DEL TRATAMIENTO CON PENTOXIFILINA Y OXIPURINOL SOBRE LA ACTIVACIÓN DE LAS MAP QUINASAS EN PÁNCREAS EN LA PANCREATITIS AGUDA EXPERIMENTAL NECRÓTICA.... 158
6.1. Efecto de la pentoxifilina y oxipurinol sobre la fosforilación de ERK en
páncreas en la pancreatitis aguda experimental necrótica. ... 158 6.2. Efecto del tratamiento con pentoxifilina, oxipurinol sobre la fosforilación de
JNK en páncreas en la pancreatitis aguda necrótica... 160 6.3. Efecto del tratamiento con pentoxifilina, oxipurinol sobre la fosforilación de
p38 en páncreas en la pancreatitis aguda necrótica... 162
7. PAPEL DEL TNF-α EN LA DEPLECIÓN DE GLUTATIÓN PANCREÁTICO EN LA PANCREATITIS AGUDA.... 164 8. EFECTO DEL TAUROCOLATO SOBRE LOS NIVELES DE GLUTATIÓN
REDUCIDO EN ACINOS PANCREÁTICOS AISLADOS. PAPEL DE LAS MAP QUINASAS Y DE LAS PROTEASAS.... 166 9. DEPLECIÓN DE GLUTATIÓN CAUSADA POR TAUROCOLATO EN CÉLULAS
ACINARES AR42J. PAPEL DE LAS MAP QUINASAS Y DE LAS PROTEASAS.. 170
9.1. Caracterización de los niveles de glutatión en células acinares AR42J. Efecto
del taurocolato. ... 170 9.2. Efecto de la inhibición de las MAP quinasas en la depleción de glutatión
producida por taurocolato en células AR42J... 173 9.3. Efecto de la inhibición de proteasas en la depleción de glutatión producida
por el taurocolato en células AR42J... 176 9.4. Papel del calcio y de la γ-glutamil transpeptidasa sobre la depleción de
9.5. Efecto del glutatión monoetil éster sobre la depleción de glutatión producida
por taurocolato en células AR42J. ... 179
10. ESTUDIO DE LA VIABILIDAD CELULAR Y DEL TIPO DE MORTALIDAD EN CÉLULAS AR42J INCUBADAS CON TAUROCOLATO.... 180
V- DISCUSIÓN ... 185
1. MODELOS EXPERIMENTALES DE PANCREATITIS AGUDA. ... 187
2. ESTRÉS OXIDATIVO Y PANCREATITIS AGUDA. ... 190
3. DEPLECIÓN DE GLUTATIÓN Y PANCREATITIS AGUDA. ... 192
3.1. Déficit de síntesis de glutatión en la pancreatitis aguda severa... 194
3.2. MAP quinasas y depleción de glutatión en la pancreatitis aguda. ... 195
3.3. Proteasas y depleción de GSH inducida por taurocolato. ... 201
3.4. Depleción de glutatión y muerte celular inducida por taurocolato. ... 204
VI- CONCLUSIONES ... 207
ÍNDICE DE FIGURAS
Figura 1.- Localización del páncreas. ... 3
Figura 2.- Teoría de la colocalización enzimática como fenómeno iniciador de la pancreatitis aguda. ... 10
Figura 3.- Papel de los diferentes mediadores de la inflamación en la fisiopatología de la PA. ... 15
Figura 4.- Síndrome de respuesta inflamatoria sistémica (SRIS)... 17
Figura 5.- Receptores de TNF-α y mecanismo de acción intracelular. ... 27
Figura 6.- Representación gráfica de los enlaces del átomo de molibdeno. ... 35
Figura 7.- Conversión de xantina deshidrogenasa en xantina oxidasa... 36
Figura 8.- Vía de degradación de las purinas. ... 37
Figura 9.- Sistemas antioxidantes celulares... 50
Figura 10.- Estructura química del glutatión reducido. ... 51
Figura 11.- Estructura química del glutatión oxidado... 52
Figura 12.- Ciclo del γ-glutamilo. ... 56
Figura 13.- Ciclo redox del glutatión... 64
Figura 14.- Via de señalización de ERK... 77
Figura 15.- Via de señalización de JNK. ... 79
Figura 16.- Via de señalización de p38. ... 82
Figura 17.- Inducción de pancreatitis con taurocolato: Material necesario. ... 108
Figura 18.- Inducción de pancreatitis con taurocolato: Preparación del animal. ... 108
Figura 19.- Inducción de pancreatitis con taurocolato: Canulación del conducto biliopancreático para la perfusión del taurocolato. ... 108
Figura 20.- Inducción de pancreatitis con taurocolato: Clampaje proximal del conducto biliopancreático para evitar el reflujo de taurocolato hacia el hígado. ... 108
Figura 22.- Canulación de la vena femoral para la administración de los
tratamientos de oxipurinol y pentoxifilina ... 108
Figura 23.- Esquema de la inducción de pancreatitis con ceruleína. ... 111
Figura 24.- Esquema de la técnica de western blotting... 126
Figura 25.- Evolución de los niveles de glutatión reducido (GSH) en páncreas
tras la inducción de pancreatitis aguda por taurocolato. ... 141
Figura 26A.- RT-PCR del gen de la subunidad catalítica de la γ-glutamil cisteína
sintetasa (GCS) en la cabeza y en la cola del páncreas... 142
Figura 26B.- Variación de la fluorescencia frente al número de ciclos de
amplificación de la RT-PCR de la GCS. ... 143
Figura 26C.- Curva de fusión de los fragmentos amplificados en la RT-PCR de la
GCS . ... 143
Figura 27A.- RT-PCR del gen de la subunidad reguladora de la GCS en la cabeza y
la cola del páncreas ... 144
Figura 27B.- Variación de la fluorescencia frente al número de ciclos de
amplificación de la RT-PCR de la GCS. ... 144
Figura 27C.- Curva de fusión de los fragmentos amplificados en la RT-PCR de la
GCS ... 144
Figura 28A.- PCR con primers específicos del exón de la subunidad catalítica de la GCS de inmunoprecipitados obtenidos utilizando anticuerpos contra
la ARN polimerasa II. ... 145
Figura 28B.- PCR con primers específicos del exón de la subunidad reguladora de la GCS de inmunoprecipitados obtenidos utilizando anticuerpos
contra la ARN polimerasa II ... 146
Figura 29A.- Unión de factores transcripcionales al promotor del gen de la
subunidad catalítica de la GCS. ... 147
Figura 29B.- Unión de factores transcripcionales al promotor del gen de la
subunidad reguladora de la GCS. ... 147
Figura 30A.- Niveles de la proteína GCS en páncreas durante la pancreatitis aguda. ... 148
Figura 30B.- Densitometría de los western blots de la GCS ... 148
Figura 31.- Niveles de glutatión reducido (GSH) en páncreas tras la inducción de
pancreatitis aguda por ceruleína en ratas.. ... 149
Figura 32.- RT-PCR del gen de la subunidad catalítica de la GCS en la cabeza del
Figura 33.- Unión de factores transcripcionales al promotor del gen de la
subunidad catalítica de la GCS. ... 151
Figura 34A.- Evolución de la fosforilación de ERK 1 y ERK 2 en páncreas durante
la pancreatitis aguda... 152
Figura 34B.- Densitometría de la cinética de la fosforilación de ERK 1 (p44) en
páncreas durante la pancreatitis aguda. ... 153
Figura 34C.- Densitometría de la cinética de la fosforilación de ERK 2 (p42) en
páncreas durante la pancreatitis aguda... 153
Figura 35A.- Evolución de la fosforilación de JNK en páncreas durante la
pancreatitis aguda... 154
Figura 35B.- Densitometría de la cinética de la fosforilación de JNK 1 (p54) en
páncreas durante la pancreatitis aguda... 155
Figura 35C.- Densitometría de la cinética de la fosforilación de JNK 2 (p46) en
páncreas durante la pancreatitis aguda... 155
Figura 36A.- Evolución de la fosforilación de p38 en páncreas durante la
pancreatitis aguda... 156
Figura 36B.- Densitometría de la cinética de la fosforilación de p38 en páncreas
durante la pancreatitis aguda... 157
Figura 37A.- Efecto del tratamiento con pentoxifilina y oxipurinol sobre la fosforilación de ERK 1/2 en páncreas a los 30 minutos de la inducción
de la pancreatitis aguda con taurocolato. ... 158
Figura 37B.- Densitometría del efecto del tratamiento con pentoxifilina y oxipurinol sobre la fosforilación de ERK 1 (p44) en páncreas a los 30 minutos de
la inducción de pancreatitis aguda con taurocolato. ... 159
Figura 37C.- Densitometría del efecto del tratamiento con pentoxifilina y oxipurinol sobre la fosforilación de ERK 2 (p42) en páncreas a los 30 minutos de
la inducción de pancreatitis aguda con taurocolato. ... 159
Figura 38A.- Efecto del tratamiento con pentoxifilina y oxipurinol sobre la fosforilación de JNK en páncreas a los 30 minutos de la inducción de
la pancreatitis aguda con taurocolato. ... 160
Figura 38B.- Densitometría del efecto del tratamiento con pentoxifilina y oxipurinol sobre la fosforilación de JNK 1 (p54) en páncreas a los 30 minutos de
la inducción de pancreatitis con taurocolato. ... 161
Figura 38C.- Densitometría del efecto del tratamiento con pentoxifilina y oxipurinol sobre la fosforilación de JNK 2 (p46) en páncreas a los 30 minutos de
Figura 39A.- Efecto del tratamiento con pentoxifilina y oxipurinol sobre la fosforilación de p38 en páncreas a los 30 minutos de la inducción de
la pancreatitis aguda con taurocolato... 162
Figura 39B.- Densitometría del efecto del tratamiento con pentoxifilina y oxipurinol sobre la fosforilación de p38 en páncreas a los 30 minutos de la
inducción de pancreatitis con taurocolato. ... 163
Figura 40.- Niveles de glutatión reducido en páncreas de ratones controles (WT) y deficientes en el receptor 1 del TNF-α (KO-RT1) y en el receptor 2 del TNF-α (KO-RT2) tras la administración de suero salino o tras la
administración de ceruleína. ... 164
Figura 41.- Niveles de glutatión reducido en ratones controles (WT) y deficientes en TNF-α (KO-TNF-α) en páncreas tras la administración de suero
salino o tras la administración de ceruleína. ... 165
Figura 42.- Efectos del taurocolato y de inhibidores de las rutas de las MAP quinasas e inhibidores de proteasas sobre los niveles de glutatión
reducido en acinos pancreáticos aislados. ... 166
Figura 43A.- Efecto del taurocolato sobre los niveles de glutatión reducido medido en células acinares de páncreas mediante microscopía confocal y su
modulación por la vía de ERK... 167
Figura 43B.- Cuantificación de los niveles de glutatión en células acinares de páncreas medidos mediante microscopía confocal utilizando monoclorobimano como fluorocromo. Efectos del taurocolato y de la
inhibición de la vía de ERK... 168
Figura 44A.- Fosforilación de ERK 1/2 en el proceso de aislamiento de acinos
pancreáticos. ... 168
Figura 44B.- Fosforilación de JNK en el proceso de aislamiento de acinos
pancreáticos. ... 169
Figura 45.- Variación del contenido de glutatión en las células AR42J tras
sucesivos pases posteriores a la descongelación de las células. ... 170
Figura 46.- Curva dosis-respuesta de los niveles de glutatión reducido en células
AR42J incubadas con taurocolato. ... 171
Figura 47.- Evolución de los niveles de glutatión reducido en células AR42J
incubadas con una concentración de 0,5% de taurocolato... 172
Figura 48.- Niveles de glutatión reducido en células AR42J incubadas durante una hora con una concentración de taurocolato de 0,5% y en
Figura 49A.- Western blot de un gel representativo de la fosforilación de ERK en células AR42J incubadas con taurocolato al 0,5% durante 1 hora y de
su inhibición mediante inhibidores específicos ... 175
Figura 49B.- Densitometría de la fosforilación de ERK 1 (izquierda) y de ERK 2 (derecha) en células AR42J incubadas con mediante taurocolato al 0,5% durante 1 hora y de su inhibición mediante inhibidores
específicos. ... 175
Figura 50.- Niveles de glutatión reducido (GSH) en células AR42J incubadas durante 1 h con taurocolato 0,5% y con diferentes inhibidores de
proteasas... 176
Figura 51.- Niveles de glutatión reducido (GSH) en células AR42J incubadas durante 1 h con taurocolato 0,5% y con inhibidores de proteasas lisosomales (monensina y 3-metiladenina) y del proteasoma
(lactacistina) ... 177
Figura 52.- Niveles de glutatión reducido en células AR42J incubadas durante 1 h con taurocolato 0,5% (Tau), con quelante de calcio (BAPTA), y con
inhibidor de γ-glutamil transpeptidasa (acivicina). ... 178
Figura 53.- Niveles de glutatión reducido en células AR42J incubadas durante una hora con taurocolato al 0,5% y con diferentes concentraciones de
glutatión monoetil éster... 179
Figura 54.- Liberación de LDH al medio de cultivo tras el tratamiento de las células AR42J con taurocolato al 0,5% durante 1 h y con el inhibidor
AEBSF... 180
Figura 55.- Cantidad de proteína en placa después de la incubación de las células AR42J con taurocolato 0,5% durante 1 h y con el inhibidor
AEBSF... 181
Figura 56.- Imagen de las células AR42J mediante microscopía confocal sin
utilizar fluorocromos... 182
Figura 57A.- Efectos del taurocolato y del AEBSF sobre los niveles de glutatión en células AR42J medidos mediante microscopía confocal utilizando
monoclorobimano como fluorocromo específico. ... 182
Figura 57B.- Efectos del taurocolato y del AEBSF sobre el tipo de muerte celular en células AR42J estudiado mediante microscopía confocal utilizando fluorocromos específicos de necrosis (ioduro de propidio)
ÍNDICE DE TABLAS
Tabla 1.- Enumeración de los criterios clínicos del SRIS. ... 13
Tabla 2.- Ligandos y receptores capaces de inducir muerte por necrosis... 89
Tabla 3.- Descripción de los anticuerpos utilizados para western blotting... 126
Tabla 4.- Condiciones de amplificación en la RT-PCR... 128
Tabla 5.- Oligonucleótidos específicos utilizados para la RT-PCR. ... 128
Tabla 6.- Anticuerpos utilizados en los ensayos de ChIP. ... 131
Tabla 7.- Condiciones de amplificación en el ChIP. ... 133
Tabla 8.- Oligonucleótidos específicos utilizados para los ensayos de ChIP. ... 133
ABREVIATURAS
- AEBSF: fluoruro de 4-(2-aminoetil)- sulfonil benceno. - AMPc: monofosfato de adenosína cíclico.
- AP-1: proteína activadora 1.
- APACHE: acute physiology and chronic health evaluation.
- Apaf-1: factor de activación de proteasas apoptóticas. - ARN Pol II: ARN polimerasa II.
- ATCC: American type cell culture.
- ATF-2: factor activador de la transcripción. - ATP: trifosfato de adenosina.
- BAPTA-AM: ester de 1,2-bis(2-amino-5-fluorofenoxy)etano-N,N,N',N'-ácido tetraacético
tetrakis (acetoximetil). - Bax: proteína X asociada a Bcl-2. - Bcl-2: proteína del linfoma de célula B 2.
- BH3: proteína con dominio homólogo a BCL-2 3.
- Bid: agonista del dominio de muerte que interacciona con Bcl-2. - Bim: mediador de muerte celular que interacciona con Bcl-2. - BSA: albúmina de suero bovino.
- CCK: colecistoquinina.
- CDNB: 1-cloro-2,4-dinitrobenceno.
- c-FLICE: enzima convertidora de interleuquina 1 beta tipo FADD. - ChIP: inmunoprecipitación de fragmentos de cromatina. - CID: coagulación intravascular diseminada.
- CK2: caseína quinasa 2. - DHLA: ácido dihidrolipoico. - DNA: ácido desoxirribonucleico. - DNAsas: desoxirribonucleasas.
- ECTA: enzima convertidora del TNF-α.
- EDTA: ácido etilendiaminotetraacético, sal disódica. - EF2: péptido de elongación 2.
- ERK: quinasa regulada por señales extracelulares. - ERN: especies reactivas de nitrógeno.
- ERO: especies reactivas de oxígeno.
- FADD: proteína asociada al dominio de muerte. - GCS: γ-glutamil cisteína sintetasa.
- GGT: γ-glutamil transpeptidasa.
- GM-CSF: factor estimulador de colonias – granulocito macrófago. - GSH: glutatión reducido.
- GSSG: glutatión oxidado.
- Hsp: proteínas de choque térmico.
- HtrA2: proteasa A2 con requerimiento alto de temperatura. - IAPs: proteínas inhibidoras de la apoptosis.
- ICAM-1: molécula de adhesión intercelular 1. - IFN-λ: interferón gamma.
- IFN-γ: interferón-γ.
- IgM : inmunoglobulina M. - IκB: inhibidor de NF-κB. - IL: interleuquina. - IP: inmunoprecipitado. - JAK: quinasa Janus.
- JNK: quinasa del NH2 terminal de c-Jun. - kD: kilodalton.
- LB: linfocitos B.
- LBA: lavado broncoalveolar. - LDH: lactato deshidrogenasa. - Linfocito TH2: linfocito T colaborador tipo 2. - LPS: lipopolisacárido.
- LT: linfocitos T.
- MAP quinasas: proteín quinasas activadas por mitógenos. - MAPKK o MEK: MAP quinasa quinasa.
- MAPKKK o MEKK: MAP quinasa quinasa quinasa.
- MCF-7: fibroblastos de adenocarcinoma mamario 7. - Mcl-1: célula de leucemia mieloide.
- MCP-1: proteína quimiotáctica de monocitos. - MDA: dialdehido malónico.
- MEF: factor estimulador de miocitos. - MEK: ERK quinasa.
- MIP: proteína inflamatoria de macrófagos.
- MKK: proteínas activadas por mitógenos quinasa quinasa. - Mo: molibdopterina.
- MPO: mieloperoxidasa. - mRNA: RNA mensajero. - NAC: N-acetilcisteína.
- NAD: dinucleótido de adenina y nicotilamida.
- NADH: forma reducida del dinucleótido de adenina y nicotilamida. - NADP: fosfato del dinucleótido de adenina y nicotilamida.
- NADPH: forma reducida del fosfato del dinucleótido de adenina y nicotilamida. - NF-κB: factor nuclear kappa B.
- NFAT: factores nucleares en células T activadas. - NGF: factor de crecimiento de los nervios. - NK: natural killer.
- PAF: péptido activador de plaquetas.
- PAK: proteína relacionada con la quinasa p21 activada. - PARS: polimerasas de poli (ADP-ribosa).
- PBS: tampón fosfato salino. - PCA: ácido perclórico. - PCR: proteína C reactiva.
- PDF: productos de degradación del fibrinógeno. - PL: plasma.
- PLA2: fosfolipasa A2.
- PMSF: fluoruro de fenil-metil-sulfonilo.
- Puma p53 up-regulate modulator of apoptosis.
- qRT-PCR: transcripción reversa - reacción en cadena de la polimerasa cuantitativa. - Ras: secuencias de DNA asociadas a retrovirus.
- RER: retículo endoplasmático rugoso. - RNA: ácido ribonucleico.
- RNApol-ChIP: inmunoprecipitación de la cromatina unida a la RNA polimerasa. - RNAsa: ribonucleasa.
- RT-PCR: transcripción reversa - reacción en cadena de la polimerasa. - SDRA: síndrome del distress respiratorio del adulto.
- SDS: dodecil sulfato sódico.
- SDS PAGE: electroforesis en gel de poliacrilamida con dodecil sulfato sódico. - SIDA: síndrome de inmunodeficiencia adquirida
- sistema ASC: sistema de transporte de aminoácidos para la alanina, serina y cisteína. - Smac/DIABLO: segundo activador de caspasas derivado de la mitocondria/ proteína de
unión directa a IAP con bajo punto isoeléctrico. - SMAD: similar to the mothers against decapentaplegic.
- SOD: superóxido dismutasa. - SP-1 proteína estimuladora 1
- SRIS: respuesta inflamatoria sistémica.
- STAT: señales de transducción y de activadores de la transcripción. - sTNF-αR receptor soluble del TNF-α.
- TAC: tomografía axial computarizada. - TBE: tampón tris-borato.
- TGF-β: factor de transformación de crecimiento β.
- TNFR: receptor del factor de necrosis tumoral α. - TNF-α: factor de necrosis tumoral α.
- TRAIL: ligando inductor de la apoptosis relacionado con TNF-α.
- tRNA: ácido ribonucleico transferente. - TxA2: tromboxano A2.
- VCAM-1: molécula de adhesión vascular 1. - XDH: xantina deshidrogenasa.
- XO: xantina oxidasa.
1. PANCREATITIS AGUDA.
1.1. Definición.
El páncreas es un órgano que muestra una longitud que varía entre 15-25 cm y que pesa entre 70 y 150 g en el adulto. Está localizado en el retroperitoneo, en íntimo contacto con otros órganos como el estómago en su porción superior, el duodeno al cual está unido, el bazo en la zona de la cola, el colon transverso por delante y el riñón y la glándula suprarrenal izquierdos (figura 1).
Ejerce una doble función, endocrina y exocrina. Se comporta como una glándula endocrina puesto que posee un 1-2% de células capaces de secretar hormonas en un páncreas adulto, la mayor parte dispuestas en los islotes de Langerhans. El páncreas endocrino está formado en un 60-75% por células β que segregan insulina y en un 20-25% por células α que segregan glucagón. El resto
son células γ y pp que segregan somatostatina y péptido pancreático
respectivamente. La mayor parte del parénquima pancreático está compuesto por células acinares que se encargan de la secreción de enzimas digestivas, que mediante conductos van a parar al intestino para ayudar a digerir los alimentos.
La diferenciación histológica y funcional de la glándula pancreática permite la clasificación de su patología según se afecte la parte endocrina o exocrina. Entre los procesos patológicos que afectan a la parte endocrina, destaca la alteración de las células β que es la causante de la diabetes mellitus. Las enfermedades que afectan la parte exocrina sobre todo son de tipo inflamatorio y tumoral. Dentro de la patología inflamatoria, por su frecuencia y repercusión, destaca la pancreatitis aguda sobre la crónica.
Los procesos inflamatorios del páncreas están desenca-denados por diversos factores etiológicos, algunos de los cuales pueden dar lugar tanto a pancreatitis aguda como crónica. La pancreatitis aguda (PA) es una enfermedad en la que el espectro clínico varía ampliamente. Mientras que en la mayoría de las ocasiones la enfermedad es leve, de curso autolimitado y mejora en pocos
días con medidas terapéuticas sintomáticas, en otras ocasiones evoluciona de forma fulminante como un cuadro de extrema gravedad y resistente a todo tipo de tratamientos. Esta variabilidad en la presentación, en la etiología y en la evolución clínica de la PA ha dificultado su estudio. Tras la primera descripción de la pancreatitis aguda por Fitz en 1889 ha habido muchos intentos de clasificación de la enfermedad, basándose al principio en criterios post-mortem o por hallazgos operatorios.
Desde 1963 se ha tratado de unificar criterios de diagnóstico, pronóstico, severidad y manejo de los pacientes con pancreatitis, realizando numerosas reuniones multidisciplinares de consenso que han incluido profesionales de todas las ramas de la salud y la medicina. Aunque con posterioridad a la reunión de Atlanta (Bradley, 1993) se han identificado defectos menores o situaciones que no quedan bien reflejadas (Dervenis, 2000; Uhl y cols., 2002), sus definiciones, criterios y clasificación siguen vigentes actualmente:
Pancreatitis aguda: proceso inflamatorio agudo del páncreas, con afectación variable de otros tejidos regionales y sistemas orgánicos alejados. Se manifiesta por la aparición súbita de dolor abdominal acompañado con frecuencia por vómitos, fiebre, taquicardia, leucocitosis, así como elevación de las enzimas pancreáticas en sangre y/u orina. Histológicamente sus hallazgos varían desde la aparición microscópica de edema intersticial y necrosis grasa del parénquima pancreático, a extensas áreas macroscópicas de necrosis y hemorragia pancreática y peripancreática.
Se diferencia la pancreatitis aguda leve de la pancreatitis aguda grave. Pancreatitis aguda leve: pancreatitis aguda asociada a una ligera alteración orgánica, con recuperación completa y ausencia de fallo multiorgánico o complicaciones sistémicas o locales. Histológicamente se presenta como edema intersticial, pudiendo encontrarse áreas microscópicas de necrosis glandular. Constituye aproximadamente el 75% de los casos de la práctica clínica y su mortalidad oscila entre el 0-5% (Beckingham y Bornman, 2001).
Pancreatitis aguda grave: pancreatitis aguda que en su evolución se asocia con la aparición de fallo multiorgánico y/o complicaciones sistémicas y/o complicaciones locales. Histológicamente se pueden apreciar áreas de necrosis y hemorragia pancreática y peripancreática, que se han relacionado directamente con la aparición de las complicaciones que van a determinar la gravedad y el pronóstico de la enfermedad(Schölmerich y cols., 1993).
Para intentar objetivar la gravedad de la PA se emplean clasificaciones pronósticas como la de Ranson que son útiles tras las 48 h del inicio de la
enfermedad (Ranson y cols., 1974) o la evaluación APACHE II que puede
McMahon, 1989). La PA grave se presenta con una frecuencia aproximada del 25% y una mortalidad que varía entre el 10-45% (Beckingham y Bornman, 2001).
Fallo multiorgánico (presencia de dos o más de los siguientes factores):
Shock: tensión arterial sistólica < 90 mm Hg.
Insuficiencia respiratoria: presión arterial de oxígeno < 60 mm Hg. Insuficiencia renal: creatinina > 177 µmol/l o 2 mg/dl tras rehidratación. Hemorragia digestiva: > 500 ml/24 horas.
Complicaciones sistémicas:
Coagulación intravascular diseminada (CID): Plaquetas < 100 000/mm3
Fibrinógeno < 80 µg/ml.
Productos de degradación del fibrinógeno (PDF) > 80 µg/ml. Alteraciones metabólicas severas:
Calcemia < 1.87 mmol/L o 7.5 mg/dl.
Complicaciones locales:
Necrosis pancreática: área local o difusa de parénquima pancreático no viable, asociado de forma característica a necrosis grasa peripancreática. La tomografía axial computarizada (TAC) con contraste intravenoso es la exploración más apropiada para su evaluación (Clavien y cols., 1988), considerándose criterio diagnóstico de necrosis la presencia de zonas bien delimitadas de parénquima pancreático no realzadas tras la administración del contraste (focales o difusas, que midan más de tres centímetros o que afecten a más del 30% del páncreas) (Balthazar y cols., 1990). La sensibilidad de esta exploración para el diagnóstico de necrosis es superior al 90%, por lo cual está considerada como el patrón de referencia para la valoración de esta complicación (Bradley y cols., 1989). Macroscópicamente se aprecian áreas de tejido pancreático desvitalizado y necrosis grasa peripancreática, bien superficiales y dispersas o bien profundas y confluentes. Microscópicamente se caracteriza por una extensa necrosis grasa intersticial, lesiones vasculares y necrosis del parénquima que afecta a las células acinares, a los islotes y a los conductos pancreáticos.
hacia su reabsorción espontánea, aunque puede progresar a pseudoquiste (si se cronifica) o a abceso (si se infecta).
Abceso pancreático: colección de pus intraabdominal, generalmente localizada en la proximidad del páncreas y que contiene poca o ninguna necrosis pancreática. Clínicamente se presenta como un proceso infeccioso tardío en el curso de la PA, habitualmente 4 semanas o más tras el inicio.
Pseudoquiste pancreático: colección de jugo pancreático limitada por una pared de tejido fibroso o de granulación. De forma ocasional se hace palpable a la exploración abdominal y su característica más importante es la presencia de una pared bien definida observable mediante ecografía o TAC. Esta complicación requiere al menos un periodo de tiempo de 4 o 6 semanas desde el inicio de la PA.
1.2. Epidemiología y etiología.
La pancreatitis aguda es una enfermedad de gran importancia debido a su frecuencia relativamente alta, y a una incidencia creciente en nuestros días. A pesar de las dificultades de ratificar los estudios epidemiológicos por las variaciones geográficas, los métodos de diagnóstico y la contabilización de brotes repetidos, se puede concluir que en los grandes hospitales entre el 0.15 y el 3% de los pacientes atendidos en urgencias sufren pancreatitis aguda (Neoptolemos y cols., 1998; Banks, 2002). De acuerdo con la Organización Mundial de Gastroenterología sobre la base de 6000 episodios de dolor abdominal agudo, el 2.3% son debidos a la enfermedad pancreática aguda y puede alcanzar el 1% de la mortalidad hospitalaria (Banks, 2002).
Es destacable que la frecuencia está aumentando durante los últimos años. En un estudio reciente que abarca 5312 pacientes en Gran Bretaña, se concluye que la incidencia se ha doblado en los últimos 30 años.
cols., 1979), hasta zonas donde se alcanzan los 370-390 casos/106 habitantes / año (Castilla-León) (Carballo, 1990). En la Comunidad Valenciana la incidencia se situaba a finales de los años ochenta en 160 casos/106 habitantes / año (Carballo y Mateos, 2002).
La etiología de la enfermedad es variada, encontrándonos con dos principales causas etiológicas, las enfermedades de origen biliar y el alcoholismo. Un 10% son debidas a causas no comunes como procedimientos quirúrgicos, infecciones, tóxicos o isquemia, y de un 10% a un 30% no se llega a encontrar la causa denominándolas pancreatitis agudas idiopáticas. En pacientes con síndrome de inmunodeficiencia adquirida (SIDA), la incidencia de la PA es mayor, bien por la propia infección vírica o bien por los efectos secundarios de la medicación antirretroviral, afectando entre el 4 y el 22% de dicha población (Dutta y cols., 1997).
Las diferencias encontradas en la etiología de la enfermedad se deben al género, edad y distribución geográfica. Así, en la mayoría de los países de Europa, España incluida, casi el 60% de los casos de pancreatitis aguda se deben a litiasis biliar, mientras que en Estados Unidos es el alcohol el principal responsable.
La edad más frecuente de aparición de la pancreatitis aguda se sitúa entre los 30 y los 70 años, con un pico a los 55 años. La distribución por sexos tiene ligeras variaciones en función de la etiología. Así, antes de los 50 años es más frecuente en varones y está en relación con la etiología alcohólica, mientras que después de esta edad es la mujer la que sufre más esta enfermedad, en relación con la mayor incidencia de litiasis biliar (Navarro, 1991). Aunque parece que el sexo masculino conlleva peor pronóstico, los resultados son contradictorios (Pezzilli y cols., 1998; Lankisch y cols., 2001).
1.3. Patogenia.
La secreción pancreática se produce de manera basal, en ausencia de comida, y se intensifica en respuesta a la comida, en la denominada secreción postprandial. Está regulada por mecanismos nerviosos, que son importantes para una regulación fina de la secreción, y con más importancia por mecanismos hormonales, destacando entre diversas hormonas que pueden intervenir la secretina y la colecistoquinina.
radica en su alcalinidad, capaz de neutralizar el quimo ácido tamponándolo para que las enzimas pancreáticas actúen correctamente. La colecistoquinina, liberada en respuesta a los productos de la digestión de grasas y proteínas, provoca la secreción de gran cantidad de enzimas digestivas por parte de las células acinares, sin aumentar de forma significativa el volumen de líquido. Estas enzimas (lipasa, amilasa, carboxipeptidasa, elastasa, tripsina y quimotripsina) son los componentes con mayor interés funcional y clínico del jugo pancreático (Navarro y cols., 2002).
La tripsina, la quimotripsina y la carboxipeptidasa son las encargadas de la degradación de proteínas y péptidos. La lipasa pancreática hidroliza los enlaces éster de los triacilglicéridos ingeridos liberando sus componentes ácidos grasos y glicerol. La amilasa actúa sobre el glucógeno, almidón y sus componentes para formar glúcidos fermentables (maltosa) y dextrinas no fermentables. Puesto que tienen tal capacidad destructiva, el páncreas debe protegerse mediante una serie de estrategias para no ser digerido. Una de estas estrategias es segregar las enzimas en formas no activas que se activarán en la luz intestinal. El tripsinógeno, forma inactiva de la tripsina, se activa por la enteroquinasa de la pared intestinal. La tripsina activa más tripsinógeno y también activa al quimotripsinógeno y la pro-carboxipeptidasa. Otra estrategia es la presencia de inhibidores de las enzimas sintetizadas por el páncreas, como es el inhibidor intrapancreático de la tripsina (α1-antitripsina).
Los ribosomas del retículo endoplasmático rugoso sintetizan las proenzimas que tras pasar a las cisternas del aparato de Golgi se agrupan en los denominados gránulos de zimógeno. Cuando se estimula la secreción pancreática, aumenta la síntesis de proenzimas y los gránulos van migrando hacia la superficie celular, descargando su contenido mediante un proceso de exocitosis en el que los lípidos y las proteínas de la membrana de los gránulos se unen a las del borde apical de la célula.
Propiciadas por las teorías de Chiari se realizaron investigaciones para evitar la autodigestión frenando o limitando la secreción de enzimas e inhibiendo enzimas pancreáticas, pero sistemáticamente los resultados inicialmente esperanzadores se transformaban en fracasos al mantenerse los efectos sistémicos de la enfermedad (Büchler y cols., 1993). Los datos experimentales muestran el papel local de la tripsina en la pancreatitis aguda. De hecho, se puede detectar la presencia de tripsina antes del daño celular acinar y también el péptido que se libera tras su activación, denominado péptido de activación del tripsinógeno (Otani y cols., 1998). Además, los pacientes con pancreatitis hereditaria presentan una mutación del tripsinógeno que da lugar a una tripsina resistente a la inactivación (Whitcomb, 1999).
En 1982, Steer y colaboradores observaron que se producía una colocalización de los gránulos de zimógeno cargados de enzimas digestivas con hidrolasas lisosomales en las fases tempranas de la pancreatitis aguda inducida por etionina (Koike y cols., 1982). La colocalización se produce mediante un proceso de crinofagia que supone la fusión de lisosomas con los gránulos de zimógeno (figura 2). Otros modelos experimentales mostraron el mismo proceso, tanto por estimulación con ceruleína como por obstrucción del conducto pancreático, llegándose a observar también fenómenos de endocitosis de enzimas secretadas y alteraciones del transporte de los gránulos de zimógeno (Watanabe y cols., 1984; Saluja y cols., 1989). Se ha demostrado que la catepsina B presente en los lisosomas es capaz de activar al tripsinógeno (Halangk y cols., 2002) y que los ratones knockout de la catepsina B presentan
menores niveles de tripsina intrapancreática tras la inducción experimental de la PA (Halangk y cols., 2000). Parece que tras la colocalización de los gránulos de zimógeno con los lisosomas, la catepsina B es la causante de la activación del tripsinógeno. Las vacuolas formadas por el mecanismo de colocalización tienen una membrana frágil pudiéndose liberar enzimas destructivas al citoplasma.
Sin embargo, diversas evidencias han cuestionado esta teoría, como la posibilidad de conseguir colocalización sin producir pancreatitis aguda (Luthen y cols., 1995b). Por otro lado, ni la administración de inhibidores de enzimas lisosomales in vivo previene el desarrollo de la PA, ni su administración in vitro
reduce los niveles de activación del tripsinógeno (Steer, 2002).
Aunque limitando la activación de tripsinógeno se disminuye el grado de necrosis acinar en los ratones knockout de catepsina B, no se consigue disminuir
Figura 2.- Teoría de la colocalización enzimática como fenómeno iniciador de
la pancreatitis aguda.
1.4. Fisiopatología. Efectos sistémicos de la pancreatitis
aguda.
La pancreatitis aguda presenta una mortalidad relativamente elevada por los efectos sistémicos que produce. Clásicamente se pensaba que el shock era la causa más importante de mortalidad inmediata (Fry, 1988), pero actualmente se considera que es el fallo respiratorio agudo, como se detalla en el apartado 1.4.2.
El shock está producido por una hipovolemia y depresión miocárdica. En las pancreatitis necróticas se aumenta el gasto cardiaco, disminuyéndose las resistencias periféricas y aumentando las resistencias pulmonares con respecto a las pancreatitis leves (Beger y cols., 1986). El íleo paralítico, los vómitos y sobre todo la formación de ascitis y extravasación de fluidos hacia el espacio extracelular, como sucede en el pulmón, produce una pérdida de volumen de aproximadamente un 30% (Wilson y Imrie, 1991). No existe afectación estructural del corazón, así que se ha asociado la posible depresión cardiaca a una disminución de la precarga o por agentes cardiacos extrínsecos (Altimari y cols., 1986) así como descenso del flujo con aumento en la permeabilidad vascular a nivel microcirculatorio, produciendo fenómenos isquémicos tanto en el páncreas como en otros órganos a distancia (Foitzik y cols., 2000). Se pueden detectar además signos de coagulación intravascular diseminada, como la elevación de los niveles de los productos de degradación del fibrinógeno, el descenso en el número de plaquetas y de fibrinógeno y aumento del tiempo de protrombina (Ohlsson y Genell, 1991).
La insuficiencia renal es una complicación frecuente, provocada por la hipovolemia y la hipoperfusión, que conduce a una necrosis tubular aguda. Menos de un 5% de los pacientes con pancreatitis aguda sufren esta complicación, pero aparece en un 20% de los que requieren cuidados intensivos. La mitad de pacientes con fallo renal agudo en la pancreatitis mueren, sufriendo mayor afectación sistémica (Ljutic y cols., 1996).
hematomas intramurales difusos, estenosis isquémicas ileales o necrosis del duodeno (Umeno y cols., 2000).
En la mitad de casos de pancreatitis aguda se produce hipocalcemia e hiperglucemia. Las zonas de necrosis secuestran el calcio por un proceso de saponificación, pero la hipocalcemia causada tiene poca repercusión clínica. La hiperglucemia probablemente sea secundaria a la destrucción de los islotes pancreáticos, a la alteración de la secreción de la insulina y del glucagón, y a la liberación de catecolaminas (Windsor y Hammodat, 2000).
Otras afectaciones menos frecuentes son la retinopatía de Purtscher (manchas blanquecinas y zonas de hemorragia alrededor de la mácula) (Hollo y cols., 1994), brotes de artritis (Hammond y Tesar, 1980), nódulos en el tejido celular subcutáneo o lesiones osteolíticas de la médula ósea por necrosis de la grasa secundaria a la liberación de la lipasa (Wilson y Imrie, 1991) e incluso encefalopatía pancreática por el paso de enzimas pancreáticas al líquido cefalorraquídeo (Boon y cols., 1991).
La enfermedad pancreática ha tenido diferentes enfoques a lo largo del tiempo. Al principio se pensaba que la mayoría de los efectos sistémicos de la enfermedad eran fruto de la liberación de enzimas por el páncreas. De hecho, la fosfolipasa A2 es capaz de alterar el surfactante pulmonar, mientras que la calicreína y la activación del complemento podrían provocar inestabilidad hemodinámica (Lasson y Ohlsson, 1984; Balldin, 1987). Ante esta perspectiva, la estrategia fue usar inhibidores de proteasas para intentar paliar los efectos de dichas proteasas activadas. Aunque se ha relacionado la cantidad de tripsinógeno y tripsina en sangre con la lesión pulmonar asociada a la pancreatitis(Hartwig y cols., 1999) los tratamientos destinados a inhibir el tripsinógeno y la tripsina no han sido del todo satisfactorios.
Diferentes estudios se centraron en la fosfolipasa A2 por su posible relación con la afectación pulmonar. La PLA2 es una enzima con una forma asociada a la membrana y otra secretoria (a su vez subdividida en dos tipos). Tiene un papel determinante en diferentes procesos inflamatorios por su acción directa sobre membranas celulares, como mediador de la síntesis de otros mediadores de la inflamación y por su acción como segundo mensajero celular (Vernon y Bell, 1992). La activación de esta enzima refleja el desencadenamiento de procesos hidrolíticos por parte de las proteasas que contienen las células pancreáticas, especialmente la tripsina.
células inflamatorias, alteración del intercambio gaseoso y alta mortalidad (Edelson y cols., 1991). Clínicamente se ha relacionado la concentración de la PLA2 con la severidad de la PA (Hietaranta y cols., 1999). De forma experimental, el aumento en los niveles de amilasa plasmática es detectable a la hora de la inducción de la PA pero los niveles de PLA2 no suben hasta las 6 h tras la inducción, lo que sugiere que la PLA2 no se libera por destrucción de las células acinares pancreáticas sino que es sintetizada posiblemente por mediadores celulares de la inflamación, macrófagos o neutrófilos (Closa y cols., 1996). A la PLA2 se le atribuyen numerosos efectos en la afectación pulmonar de la PA, la activación de los neutrófilos y el incremento en la producción del péptido activador de plaquetas (PAF) y del óxido nítrico en los pulmones (Tsukahara y cols., 1999).
1.4.1. Síndrome de respuesta inflamatoria sistémica (SRIS).
El fracaso de los tratamientos destinados a inhibir la fosfolipasa A2 y las pocas diferencias que existen entre el síndrome de respuesta inflamatoria sistémica que aparece en la PA y el que surge en otras enfermedades, como puede ser el shock séptico o los grandes quemados, han provocado que se deba cambiar la visión y no nos limitemos sólo a enzimas activadas. Rinderknecht a finales de los años ochenta (Rinderknecht, 1988), propone que son los mediadores de la inflamación los que provocan la activación de las células inmunológicas en órganos a distancia, y que la sobreestimulación o la respuesta incontrolada de estas células daría lugar a las lesiones tisulares que harían fracasar los diferentes órganos hasta entrar en el fracaso multisistémico. De esta forma, serían los mediadores de la inflamación el nexo de unión entre la lesión pancreática, proceso aparentemente local, y la lesión de órganos a distancia que hacen de este proceso una enfermedad sistémica (figura 3).
Tabla 1.- Enumeración de los criterios clínicos del SRIS.
CRITERIOS CLÍNICOS DEL SRIS (se deben cumplir dos o más)
Temperatura > 38º C o < 36º C.
Frecuencia cardiaca > 90 latidos / minuto.
Frecuencia respiratoria > 20 respiraciones / minuto.
Actualmente, la tendencia es a considerar la pancreatitis aguda grave como bifásica. La primera fase se corresponde con un síndrome de respuesta inflamatoria sistémica (SRIS) que conduce al fallo multiorgánico responsable de la mortalidad inicial, que puede alcanzar el 60% de los pacientes fallecidos por PA, especialmente cuando existe afectación pulmonar. Si fallan los mecanismos de defensa natural o falla la intervención terapéutica, se desarrollará una segunda fase caracterizada por la aparición de complicaciones locales, generalmente de tipo séptico, que van a crear un segundo pico de mortalidad (Neoptolemos y cols., 1998).
Se considera que el SRIS surge por el descontrol de la respuesta inflamatoria necesaria del organismo ante determinadas agresiones que, en condiciones normales, quedaría confinada al órgano de origen por la acción de inhibidores, agentes antiinflamatorios o mecanismos de contrarregulación. Participan en el SRIS los denominados mediadores de la inflamación los cuales pueden ser clasificados en: componentes celulares, (sobre todo macrófagos y neutrófilos), mediadores de origen celular y mediadores de origen humoral (figura 4). La pancreatitis aguda es por tanto un modelo ideal para estudiar estos procesos de inflamación sistémica (Wilson y cols., 1998).
El fracaso multiorgánico hace referencia a la evolución del SRIS, y se define como el síndrome progresivo, potencialmente reversible, de fallo orgánico que afecta a dos o más sistemas diferentes del órgano de origen. Los síntomas pueden variar entre una disfunción leve transitoria y el fallo irreversible de un órgano, siendo la mortalidad directamente proporcional a la rapidez de instauración y al número de órganos afectados (Giroir, 1999; Buter y cols., 2002).
El grupo de Norman (Norman y cols., 1995a), mediante técnicas de inmunohistoquímica y determinación de mRNA, ha puesto de manifiesto que tras la inducción de la pancreatitis aguda los macrófagos son los iniciadores de la respuesta inflamatoria y, por su capacidad de activar otros leucocitos y producir grandes cantidades de mediadores proinflamatorios, extienden y amplifican la inflamación. Estudios in vitro de monocitos obtenidos de pacientes con
Figura 3.- Papel de los diferentes mediadores de la inflamación en la fisiopatología de la PA.
Macrófagos Neutrófilos Linfocitos Hígado Inflamación pulmonar TNF-α TNF-α TNF-α IL-1β IL-1β IL-6 XO XO PAF PAF PAF
FALLO MULTIORGÁNICO
TNF-α IL-1β XO TNF-α IL-1β XO Proteasas TNF-α IL-6 XOTNF-α PAF IL-8
ROS
TNF-α
NO
PAF
ROS
LESIÓN CELULA ACINAR
Macrófagos Neutrófilos Linfocitos Hígado Inflamación pulmonar TNF-α TNF-α TNF-α IL-1β IL-1β IL-6 XO XO PAF PAF PAF
FALLO MULTIORGÁNICO
TNF-α IL-1β XO TNF-α IL-1β XO Proteasas TNF-α IL-6 XOTNF-α PAF IL-8
ROS
TNF-α
NO
PAF
ROS
La implicación de los neutrófilos en la fisiopatología de la PA ha quedado demostrada tanto por estudios morfológicos como por la presencia de productos derivados de los neutrófilos en el páncreas, en el pulmón y en el plasma de estos pacientes. Un ejemplo es la mieloperoxidasa, enzima que contienen los neutrófilos, que se utiliza como marcador de infiltración inflamatoria tisular por neutrófilos (Folch y Closa, 2001). La tripsina puede hidrolizar proteínas del complemento produciendo los agentes quimiotácticos C3a y C5a, que podrían reclutar neutrófilos en el páncreas (Acioli y cols., 1997). La elastasa también está implicada en el reclutamiento de neutrófilos (Tsuji y cols., 1994). En modelos de pancreatitis edematosa estos productos parecen ser la principal fuente de radicales libres (Closa y cols., 1994).
La adhesión de los neutrófilos al endotelio y la migración a los tejidos inflamados está controlada en parte por quimioquinas. Se produce en cuatro
pasos definidos; rodamiento, activación, fuerte adhesión mediada por integrinas y por último, migración mediante diapédesis.
Figura 4.- Síndrome de respuesta inflamatoria sistémica (SRIS).
Dentro de los componentes de origen celular, cabe destacar las citoquinas y los radicales libres que se desarrollarán en los siguientes apartados. Asimismo hay que mencionar el factor activador de plaquetas (PAF) y los eicosanoides. La regulación de la producción de PAF está íntimamente relacionada con IL-1 y
ESTÍMULO
Activación Neutrófilo Activación Plaquetar Metabolismo Ácido AraquidónicoAdhesión Leucocitaria, Inflamación Sistémica, Activación Endotelial
Daño Endotelial, Trombosis Intravascular, Vasodilatación, Hipotensión, Depresión
Miocárdica, Distress Respiratorio
DISFUNCIÓN MULTIORGÁNICA
IL-1, IL-8
IL-8 PAF
Complemento Sist coagulación Proteínas fase aguda
Estrès
oxidativo NO
PLA2 Elastasa
TNF-α IL-1
PAF IL-8
LTC4 TxA2
PgE2 ICAM VCAM Quininas PAF TNF IL-6
Adhesión Leucocitaria, Inflamación Sistémica, Activación Endotelial
Daño Endotelial, Trombosis Intravascular, Vasodilatación, Hipotensión, Depresión
Miocárdica, Distrés Respiratorio
FALLO MULTIORGÁNICO
IL-IL-8 PAF
Estrès
oxidativo NO
PLA2 Elastasa
TNF-α IL-1
PAF IL-8
LTC4 TxA2
PgE2 ICAM VCAM Quininas PAF TNF IL-6 Degranulación Mastocito
Proliferación L T
Estimulación L B
Activación Macrófago
ESTÍMULO
Activación Neutrófilo Activación Plaquetar Metabolismo Ácido AraquidónicoAdhesión Leucocitaria, Inflamación Sistémica, Activación Endotelial
Daño Endotelial, Trombosis Intravascular, Vasodilatación, Hipotensión, Depresión
Miocárdica, Distress Respiratorio
DISFUNCIÓN MULTIORGÁNICA
IL-1, IL-8
IL-8 PAF
Complemento Sist coagulación Proteínas fase aguda
Estrès
oxidativo NO
PLA2 Elastasa
TNF-α IL-1
PAF IL-8
LTC4 TxA2
PgE2 ICAM VCAM Quininas PAF TNF IL-6
Adhesión Leucocitaria, Inflamación Sistémica, Activación Endotelial
Daño Endotelial, Trombosis Intravascular, Vasodilatación, Hipotensión, Depresión
Miocárdica, Distrés Respiratorio
FALLO MULTIORGÁNICO
IL-IL-8 PAF
Estrès
oxidativo NO
PLA2 Elastasa
TNF-α IL-1
PAF IL-8
LTC4 TxA2
PgE2 ICAM VCAM Quininas PAF TNF IL-6 Degranulación Mastocito
Proliferación L T
Estimulación L B
TNF-α, puesto que la inhibición de la producción de cualquiera de ambas citoquinas atenúa la producción de PAF, mientras que la inhibición de PAF atenúa la producción de IL-1 y TNF-α (Norman, 1998). De forma experimental, se ha mostrado que produce una liberación masiva de amilasa por parte de las células acinares, promoviendo el infiltrado inflamatorio del páncreas. El páncreas es capaz de producir PAF y las concentraciones pancreáticas aumentan en la pancreatitis aguda (Zhou y cols., 1993). También aumenta en pulmón y en sangre en el desarrollo de la enfermedad (Zhou y cols., 1993). La administración intraperitoneal o intravascular de PAF en modelos experimentales puede provocar o incrementar la severidad de la pancreatitis aguda. Por este motivo se han llevado a cabo estudios clínicos y experimentales con inhibidores del PAF (como el lexipafán), que demuestran una disminución de la respuesta inflamatoria sistémica (Johnson y cols., 2001).
Por su parte, el papel de los eicosanoides en la pancreatitis aguda podría ser algo contradictorio, puesto que en los diferentes modelos experimentales se observan variaciones en los niveles plasmáticos y en los niveles tisulares de cada metabolito. No obstante, algunos leucotrienos son potentes estímulos quimiotácticos para los neutrófilos y la inhibición de la ciclooxigenasa disminuye la severidad de algunos modelos de PA (Ethridge y cols., 2002). Parece que es más importante el papel citoprotector de las prostaglandinas. Estas moléculas son capaces de estabilizar las membranas, evitan la formación de enzimas capaces de generar radicales libres derivados del oxígeno y mejoran el flujo sanguíneo pancreático (Hirano y cols., 1992; Closa y cols., 1993).
Es importante destacar la fracción C5a del sistema del complemento. Esta fracción es un potente mediador proinflamatorio que estimula la contracción del músculo liso e incrementa el flujo vascular y la permeabilidad capilar y atrae los neutrófilos a los pulmones. Sin embargo, se han obtenido algunos resultados paradójicos ya que su inhibición parece agravar la severidad de la PA y la lesión pulmonar asociada (posiblemente porque C5a estimula también la liberación de la citoquina antiinflamatoria IL-10), lo que podría sugerir un papel contrarregulador del sistema del complemento o de alguna de sus fracciones en el SRIS producido por la PA (Saluja y Steer, 1999).