UNIVERSIDAD A U T ~ N O M A METROPOLITANA
UNIDAD IZTAPALAPA
DIVISIÓN DE CIENCIAS BÁSICAS E INGENIERíA
DEPARTAMENTO DE QUÍMICA
ÁREA DE QU~MICA CUÁNTICA
"SIMULACIÓN DE
MEZCLAS
BINARIASEN EL EQUILIBRIO LÍQUIDO-VAPOR "
TESIS QUE PRESENTA EL ALUMNO:
FRANCISCO
NOÉ
MENDOZA AMBROSIO92221 1 IS
PARA LA O B T E N C I ~ N DEL GRADO DE:
Q U ~ M I C O
ASESOR:
DR. JOSÉ R. ALEJANDRE RAMíREZ
ESTE PROYECTO DE INVESTIGACIÓN
SE
REALIZO
EN EL ÁREA
DE
QU~MICA
CUÁNTICA, DEL DEPARTAMENTO DE
QUÍMICA
IZTAPALAPA.
DE
L.A
UNIVERSIDAD
A U T ~ N O M A
METROPOLITANA, UNIDAD
BAJO
LA
D I R E C C I ~ N
Y ASESORÍA DE:
~
DR. JOSÉ R. ALEJANDRE RAMíREZ
LO PRESENTA :
FRANCISCO NOÉ MENDOZA AMBROSIO
AGRADECIMIENTOS
Agradezco al Instituto Mexicano del Petróleo y a la Universidad Autónoma Metropolitana
unidad Iztapalapa
por su ayuda y las facilidades para realizar este proyecto de investigación.
Quiero agradecer a mi familia el apoyo y confianza que me brindaron
Este trabajo tuvo el soporte y el apoyo de mi asesor, Dr. José R. Alejandre, a quien agradezco
la paciencia que me tuvo, y la ayuda que me prestó
Especialmente quiero agradecer a mi padre Heladio, a mi madre Rosita, a mi hermana Alia María, u mis hermanos
INDICE
RESUMEN
INTRODUCCI~N
POTENCIAL DE INTERACCI~N
DINÁMICA MOLECULAR
PROPIEDADES
RESULTADOS
TENSIÓN SUPERFICIAL
CONCLUSIONES
BIBLIOGRAFIA
4
1
10
17
30
RESUMEN
Simulaciones de dinámica molecular a temperatura constante son
realizadas en la interfase líquido-vapor de mezclas binarias. Se
eligieron parámetros de potencial para validar el método de simulación con resultados en la literatura. Se obtuvieron
deiisidades ortobáricas, presión y tensión superficial como
funciones de la composición. Nuestras curvas de coexistencia
difieren de las reportadas debido a la dificultad de incluir en
nuestro método las correcciones de largo alcance en las
interacciones. Nuestros resultados de la presión de vapor son
mayores que en trabajos anteriores. Nuestros resultados tienen la
misma tendencia que los observados en la literatura. Nuestro método, a diferencia de otros, permite calcular la tensión
superficial.
Los
métodos de simulación requieren de las interacciones moleculares para la obtenciónde propiedades macroscópicas de interés experimental (diagramas de fases, propiedades
termodinámicas, etc.), hay dos maneras de calcular estas propiedades, con métodos
probabilísticos (montecarlo)
y
con métodos determinísticos (dinámica molecular)3Desde el punto de vista de la simulación hay diferentes métodos para estudiar el equilibrio
entre fases. Hay métodos donde la interfase no esta presente como: la integración
Termodinámicai, la integración de Gibbs-Duhem2, y el método del ensamble de GibbsX.1, con este último método hicimos nuestras comparaciones de propiedades, dado que es un método aceptado y generalizado. Nosotros usamos u n método deterministico conocido como Dinámica Molecular, donde la interfase esta presente y podemos calcular
sus propiedades. Los métodos con o sin interfase producen resultados equivalentes para
el miqmo modelo de potencial.
El objetivo de este trabajo es desarrollar la metodología para estudiar propiedades
interfaciales de mezclas binarias quc interaccioiian con el potencial de Lennard Jones y
obtener las curvas de coexistencia y la tensión siiperficial cn función de la concentración
dc uno de los compuestos. Las simulaciones se hacen a temperatura constante .Los
sistemas se cligieron para comparar los resultados con los reportados en la literatura.
Se debe notar que el potencial de interacción usado en este trabajo es esféricamente truncado, mientras que en la literatura se reportan los resultados para el potencial
completo. Para disminuir las diferencias usamos un radio de corte grande, sin embargo
eii algunas mezclas esto no fue suficiente.
En las secciones siguientes se describe cl potencial, la metodología, y los resultados
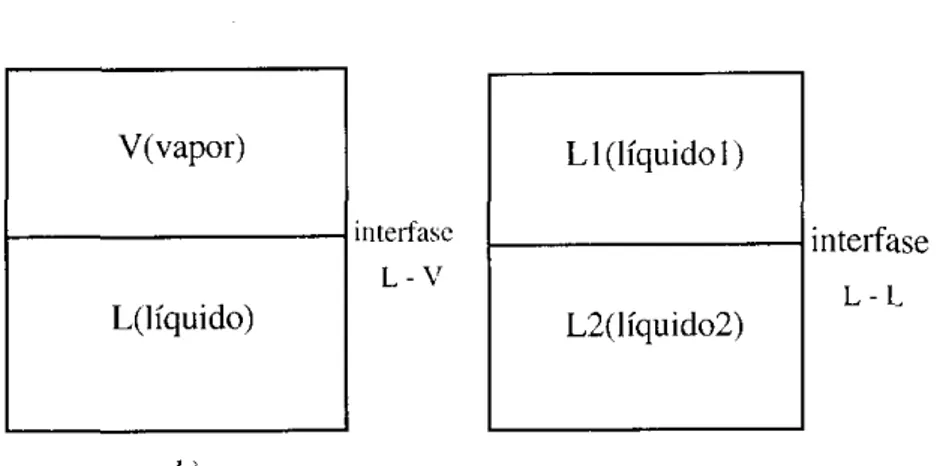
obtenidos en este trabajo. En la figura I se muestran los dos tipos de interfase niás
comunes.
V(vapor)
iriterfasc L -
v
L(líquido)LI (Iíquidol)
-1
interfaseL - t L2( Iíquido2)
b)
POTENCIAL DE INTERACCI~N
El potencial de interacción de Lennard-Jones se uso para simular mezclas binarias y se define de la siguiente forma,
donde
rii
es la distancia entre dos moléculas, es la profundidad del pozo de potencialentre las moléculas de la especie
ap,
yap
es el diámetro de las moléculas. Paracomparar nuesíros resultados hicimos las simulaciones para tres tipos de mezclas, que se
definen con los siguientes parámetrosx.
MEZCLA E l l E 1 2 l. 22 0 1 I o 1 2 o 2 2
I. 1 0.75 I 1 I 1
I1 1 1 I 1 0.885 0.769
111 I 0.773 0.597 I 0.884 0.768
Tabla I . Pdrámetros del putenrial en la simulaciún de las
tres mczclas.
La mezcla I tiene parámetros para simular un sistema parcialmente miscible, la mezcla I1 es totalmente miscible y solo se cambia el tamaño de las especies, la mezcla
I11
estotalinente miscible, y se cambia el tamaño de las cspecies y la magnitud de la interacción
entre ellos.
DINÁMICA MOLECULAR
Antes de comenzar la simulación debemos de conocer, además del potencial de
interacción la cofiguración inicial. la posición y la velocidad de la moléculas,
Para hacer la dinámica molecular es necesario resolver la ecuación diferencial de Newton del movimiento para un número N de partículas con respecto al tiempo.
donde
F,
es la fuerza sobre el átomoi
debida a las interacciones con los demás átomos del sistema.
m, es su masa y u, es su aceleración.Usando algoritmos de integración cn el tiempo de la ecuación de movimiento de los
átomos que interactúan con un potencial molecular, podemos seguir su trayectoria. Estos
algoritmos se obtienen con expansiones truncadas de la serie de Taylor de la posición de las partículas alrededor de un tiempo I,
Sumando las ecuaciones 4, 5 y truncando hasta la potencia de orden cuarto se obtiene,
restando las mismas ecuaciones y truncando hasta la potencia de orden tres se obtiene,
cl algoritmo de veriet’
v,(t)=
r i ( t + A f ) - r i ( t - A f )2Af (7)
Podemos predecir las posiciones de los átomos al tiempo t
+
Af y realizando variasconfiguraciones con este procedimiento se puede determinar la trayectoria de las moléculas y obtener propiedades temodinámicas tales como: temperatura, presión,
densidad, tensión superficial, etc. Para garantizar que tenemos un sistema infinito usamos
condiciones periódicas en las tres direcciones, como se muestra en la figura 2 para un
sistema bidimensional.
.
I .X
.. .
.
..
e .--"-
Figura 2. Mucstm una i d a de la ccld;i unilaria
y de la pcriodicidad en iI plano Y ~ X
La celda se llama celda unitaria y sus dimensiones se determinan con la densidad del
sistema inicial y el número de partículas. Las variables que definen el sistema son N, V , T y se inantienen constantes durante la simulación. La manera de construir la interfase se
ilustra en la figura 3, se divide una celda rectangular en tres zonas en la dirección Z . AI
inicio todas las moléculas se encuentran en la celda central y conforme se desarrolla la
siinulacibn, las moléculas se difunden a las otras zonas.
rnponente A
a
6 0 80C o o r d e n a d a en 2
figura 3. Mucstra cornu se construye la celda de simulaciún y l a región de la intertáse.
Nosot.ros hicimos simulaciones de Dinámica Molecular en el ensamble NVT usando
condiciones periódicas en las tres direcciones de una celda paralelepipeda. Se usaron
unidades reducidas en términos del radio de las moléculas, y E . El incremento de tiempo
reducido fue de 0.005. L a simulación se realiza truncando la fuerza a 4.4 veces el
diámetro de las moléculas . El uso de este truncamiento o radio de corte es para obtener
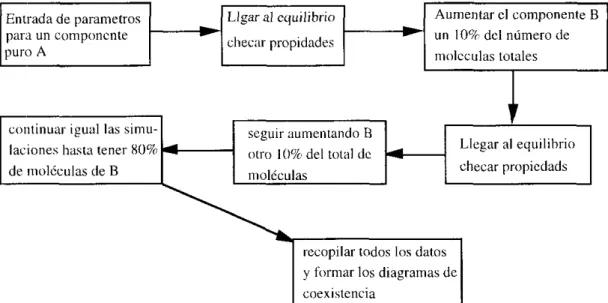
un potencial muy parecido al completo. Después de 100,000 ciclos de simulación teniendo en cuenta que un ciclo es un paso de tiempo reducido, el sistema se equilibra y las propiedades promedio se calcularon con otros 400,000 pasos de tiempo. En la figura 4 mostramos un esquema de la forma de proceder.
Entrada d e parameiros para un componcnie puro A
-
Llgar ill equilihrio
clieciir propidades
Aumentar el componente B
un 10% del número de
iiidcculas totales
-
Figlira 4. Alguriinio rcguiilo para el dcsarroll<i de la
sirnulaci6n. continuar igual las simu-
laciones hasta tener XU%
d e iiio1Cculas de B
En la siguiente tabla damos los parametros empleados
scguir aumentando B
iiioléculas
Llegar al equilihrio checar propicdads
4 otro ¡(I%' del t«ial dc 1
# de moléculas
totales radio de corte
mezcla Temperatura
I 1.15 2216 4.4
I1 1.15 2216 4.4
111 0.928 2000 4.4
tabla 2. Se inueriran Ius I>aráiiicir«s para cada i i i c ~ c l a se cligiernn así para piidcr cuinparar 1x1.
PROPIEDADES
El procedimiento usado para calcular las propiedades de la interfase es el mismo que se
usa para el potencial de Lennard-Jones en la interfase Iíquido-vapot4 de un componente. Las densidades de coexistencia, son obtenidas al final de la simulación, por el ajuste del promedio de perfiles de densidad a una función tangente hiperbólica.
donde
p,,
ype,
son las densidades del líquido y del vapor en el bulto, zo es la posición de Gibbs que divide la interfase y d indica el espesor de la interfrise. En la figura 4 se muestra el perfil de densidades que se calcula de la siguiente manera.*
0.6 N0.2
O
t
I
I
t
v a o o r ~ v BI
i
densidad del liquido B
O 20 40 60 BO
coordenada " z a r
*
Unidades Reducidasfigura 5. Se niucstra el pei-lil de dcrisidadcs de una nieiclil hinaria miscihleimczc1;i 111)
Se divide la celda en N rebanadas sobre el eje Z, de espesor Az y se encuentra el número
de moléculas en cada rebanada, donde
AV
es el volumen de una rebanada, N es el número de moléculas en esa rebanada.La curva ortohárica es obtenida con simulación a diferentes temperaturas. La presión normal y tangencia1 en el plano interfacial son calculadas usando la definición de Irving y
Kirkwoods.
Donde p ( z ) es el perfil de densidad promedio, i: es la función Heaviside, A es el área
interf;cial, k,, es la constante de boltzinann, T es la temperatura y es la derivada
del potencial. En esta expresión el primer término corresponde a la contribución de la
energía cinética, mientras que el segundo es debido a las fuerzas intermoleculares. Los valores que la función Heaviside puede tomar son: Q(z) = O si
z
<
O y@(z)
=Isi
z
;.
o, En la figura seis se muestra la diferencia de presiones normal y tangencial.-0.1 I I I
¿
0 20 40 60 a0
coordenada en Z
figura h. La diferencia de la presión normal y tangencia1
cn funcióii de la iiosiciiin de las riioléciilas eii la dirección z
Para un sistema denso y u n potencial molecular de largo alcance el último termino nos
proporciona la contribución más importante La presión en el vapor y en la fase líquida es igual a la presión normal. L a presión normal y tangencial son iguales en la fase líquida y
en la fase vapor, como puede verse en la figura. La definición de la tensión superficial es:
Donde el factor de 1/2 se toma porque hay dos interfases en el sistema simulado
-0.3
~-0.5 I I I I
20 40 60 80
O
coordenada en Z
figura 7 . Se muestra gráficamente la integral
de la tensión supcrliciiil,
En la figura 7 se observa un perfil de la tensión superficial, en unidades reducidas. En la
fase vapor y líquida la contribución a la tensión superficial es aproximadamente cero.
RESULTADOS
Con los parámetros de las mezclas hicimos simulaciones a temperatura constante y
obtuvimos los siguientes datos para cada tipo de mezcla.
para la mezcla 111 T=0.928
0,5925
1800 0,4023 1600 0,3180 1400 0,2241 1200 0,1826 I000 0,1412 800 0,1200 600 0,1 136 400200 0.03 0.353 400 0.0433 0.0506 600 0.0560 0.0672 800 0.0799 O. 1036 1 000 0.1033 0.1400 1200 0. 13.53 0. 1920 I400 0.1614 0.2519 I600 0.2406 0.5748
0.7452 0.4790 0.7682 0.4276 0.7895 0.4124 0.8208 0.3063 0.8454 0.2446 0.874 I 0. 1888 0.8778 0.0710 0.47 I6 -0.0020
para1;irnczclaII T=I.IS
0.9398 21 16 100 0.07 19 0.0909 0.594 1 0.1431 0.7804 1816 400 0.0798 0.0977 0.6533 0. 16 I8 0.6702 1616 600 0.082 I 0.0982 0.6896 0. 17 10 0.5657 1416 800 0.0882 O. 1 056 0.7382 0. 1805 0.4803 ~ ~~~~ 1216 ~~~~ ~~~~ 1000 0.09 16 ~~ ~ 0. 1069 ~ ~~~ 0.784 I ~ ~~ 0.2 ~ ~~~~ 122
0.3572 1016 1200 0.0977 0.1 151 0.83.57 0.2214 0.2672 816 1400 0. 1 I 05 0. 1326 0.9 1 13 0.2226 0.2060 616 1600 0. I 1.54 0.1442 0.9802 0.2640 0.1404 416 1800 0. 1224 0. I468 1.0700 0.2652 0.0629 216 2000 0. 1360 0. I64 I I . I797 0.2846
paralamezcla1 T=1.15
0.0898 I O 0 2116 0.0367 0.0386 0.6904 0.3412 0.2679 200 2016 0.0430 0.0533 0.6686 0.6329 0.3660 300 1916 0.0470 0.0592 0.6652 0.2713 0.4234 400 1816 0.0490 0.0605 0.6550 0.2358 0.4427 500 1716 0.0527 0.0689 0.6501 0.1946
iahla 3. Se dan los resiiliados de ;ilgun;is prupiedades
unidades ridiicidas
q u i cc C"c«ntiallln C o n la simuiac,ó,,. l<id<i cstu cn
donde:
X,l,l,
XA,,,
son las fracciones molares del componente A en la fase líquida y en la fase vapor, respectivamente y nA, nB son el núinero de moléculas del componente A y del B,P,
es la presión de vapor total. ( P , ~+pll)(V)
es la suma de las densidades en el vapor del componente A y el componente B, (P,~ +p,)(l) es la suma de las densidades del componente A y del componente B en la fase líquida,y
es la tensión superficial de lamezcla.
Con los datos de la tabla 3 se construyeron las figuras de presión de vapor vs fracción
molar para los tres tipos de mezclas, también se incluyen los perfiles de densidad para los puntos A y B en cada uno de los tipos de mezclas
0.070
-
-
< . e -
_ I _,-- -
,- .-
.
.
-9.. ..
,-
_ _ * - VAPOR-
_ -
- :,.
'.
I-Punto A
0.2
J
1
Comp. B
w
O 20 40 60 80
C o o r d e n a d a en
Z
Figura Y. Se muestra el [icrfil de densidades para el punto A
del diagrama de coexistencia de la mezcla I con SO0 moléculas de la especie A y 1716 de 11 especie B.
Punto B
0.8
-
-
?
-'O.
6
L
L
*
-
N vC o o r d e n a d a en Z
Figura IO. Sc ~mucstra cI prrfil dc densidades para 51
con 100 ~mol~culas de I:I cipccir A y 21 16 de la punto H del diagrama <IC ci>existeilci;i de lil lllc7~cl;l I,
Para la mezcla I1 se hicieron las siguientes curvas de coexistencia y de perfiles de
densidad
0.1 4
0.1 2 *
L
O
Q
U
>
o.
1u c
O
m
a\
L a
._
0.08
Punto A
-
nuedrar
+--
- + referencia [XI-\
-
nuedrar+---+referencia [XI
VAPOR
I I I I I I I I I
O
0.20.4
0.6 0.8 10.06
Fraccion molar X(A)
Figura I I , Presión de vapor contra composición
para la rnczcla II, comparados con los de la referencia(x1.
en unidades reducidas.
Punto
A
1.5
-
-
1 - -
- -
0.5
-
-
-
-20 40
O O
C o o r d e n a d a en Z
Figura 12. Perfil de densidad par;, el punto A
de la rne~cla II. c m 216 mol6culas del Iipo A
y 2000 moléculas de R.
Punto
B
I
I I I I I I
Comp. B
1
O 20 40 60
O
C o o r d e n a d a en Z
I
1
OFigura 13. Pcrfil d e densidad pare cI punlo I3 de la mezcla I¡. con 21 16 moléculas dc A y 100 moléculas
de B
0.30
I I I I II
I I I4
0.25
L
1
*
L
O
E
0.20
>
c
ai
0.15
i
I-
O
._
z
L 0.10 a
0.05
1
)--I nuestros Punio
-
referencia[x]LIQUIDO
VAPOR
0.00
0.20
0.40 0.600.80
1
.o00.00
fraccion molar X(B)
Figura 14. Diagrama de cocnistencia liquido- vapor para la mezcla 111 con la fracción del componente B en CI j c "X" coinparddoS con l a refcrencia(X).
Punto A
0.8 KS I I I I I I I
- Componente A
CI
-
0.6 -
-
*-
v N - -
77
0.4 -
-
77
a v1 tz
a> ~
~
0.2 -
-
- Componente
B
-C o o r d e n a d a en Z
Figura 15. Perfil de densiikad pera el punto A del diagrama de coexistencia de la mricla 111 con 1800 inoléculas del tipo A y 200 moléculas dcl iipo B
Punto
B
0.2
O - - --
-O
10
20
30
40
50
Coordenada en Z
Figura 16. Perfil de densidades par a c1 punio B dcl diagrama dc coexistencia de la mezcla 111, con 400 molkculas dcl iipo
A y 1600 moléculas del iipo R. En este csiado el sistema comienza a ser homogineo
TENSIÓN SUPERFICIAL
SC muestran en la tabla siguiente, resultados de tensión superficial para los diferentes
tipos de mezclas que se simularon
pira la mezcla 1
XA,I
Y
0.1 1 IS 0.0965 0.0750 0.0700 0.0466
o para al mezcla I1
0.1415 O. 1586 0.1618 0. I687 0. 1959 0.1951 0.2165 0.2477 0.2486 0.2995
para la mezcla 111
0 ,3822 O .3760 0 ,3458 0 ,3262 0 ,2161 0 ,1300
o
,1200 O .O700 O ,0130 O ,0039donde XAJ es la fracción mol del componente A en el líquido,
y
es la tensión superficialde la mezcla.
Las gráficas que a continuación se muestran son resultado de la simulación en
dinámica molecular que nosotros obtuvimos, la tensión superficial esta en unidades
reducidas. y* I= y-
o*
&
*
F
I I I I I
fraccion mol en el liquido X(A)
figura 17. Tcniióii supcrficial YS fracción inol
del componcnie A en el líquidii para la rnc~cla I a T=l. 15
tensión superficial para la mczcla
I1
a
I I I I I I I I I 0.30 -.tensión superficial para la mczcla
I1
a
I I I I I I I I I 0.30 -.-
*
U
U
.-
0.25 -.-
L
W
Q
v> 0.20 -
c O v> L W
.-
u 0.15 -
t
I I I I I I I I I
0.0 0.2 0.4 0.6 0.8
0.1
o
fraccion mol e n el liquido
X(A)
Figura 18. Tcnsiún supcrficial V E friiccióii nid
del comp«nentc A ell ií<luido iii Tniicia II ~ T = I . I S
I
I O
Tensión superficial para la mezcla
1110.40
4
I I I I I I I I.L?
0.30
v
.-
LL
a>
Q
2
0.20c
o
3 0.10
.-
(B
c
0.00
t
I
i
I
I
I I I I IU.0 0.2 0.4 0.6 0.8 1
.o
fraccion mol en el
liquido
X(A)
Figura 19. Gráfica de tensión supdicial vs fracción mol dcl componente A en el líquido para la mezcla 111 u T=0.928
CONCLUSIONES
Los resultados para la curva de coexistencia tienen la misma tendencia que los reportados
en la literatura.
En
nuestro caso la presión de vapor es sistemáticamente mayor debido a la no inclusión de las correcciones de largo alcance en el potencial. Este efecto es menor enla inezcla 111, debido a que uno de los componentes tiene
inenoi'
tamaño y menor pozo de potencial.L a metodología desarrollada permite estudiar propiedades de coexistencia e interfaciales de mezclas binarias en el equilibrio líquido-vapor.
Actualmente estamos desarrollando un método para incluir la interacción completa del potencial. El método lo aplicaremos al estudio e interfases de sistemas reales.
Bibliografía
I) Gubbins Keith E. Molcular Simulation, 1989, V01.2, pag. 223-252
2) Mehta Manoj and Kofke David D., Chemical Engineering Science, 1994, Vo1.49, No.16, pag.2633-2645
3) Allen M.P. and Tildesley D.J., Computer Simulation of liquids, Oxford University Press, Oxford, 1987
4) Alejandre J., and Tildesley D.J.,J. Chem. Phys. 1995, Vo1.102, pag.4574
5) Irving
J.H.
and KirkwoodJ.G.,
J. Chem. Phys., 1950, Vo1.18, pag.817 x) Panagiotopoulos A.Z., Quirke N., Stapletoii M., Tildesley D.J., MolecularPhysics, 1988, V01.63, No.4, pag.527-545