INSTITUTO POLITÉCNICO NACIONAL
ESCUELA SUPERIOR DE INGENIERÍA MECÁNICA Y ELÉCTRICAUNIDAD PROFESIONAL “ADOLFO LÓPEZ MATEOS”
TESIS
“PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE
CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA”
QUE PARA OBTENER EL TÍTULO DE
INGENIERO EN CONTROL Y AUTOMATIZACIÓN
PRESENTA
VANESSA JAZMÍN GONZÁLEZ CUÉLLAR
ASESORES: ING. ARTURO ROLANDO ROJÁS SALGADO ING. JOSÉ ANTONIO MARTÍNEZ HERNÁNDEZ
AGRADECIMIENTOS
A mis PADRES, María de La Luz Cuéllar y Roberto González por darme la vida, por verme crecer y cuidarme, por haber sembrado en mí esa semillita de seguir estudiando, prepararme y terminar la Carrera, ayudarme con el sustento de mis estudios, por estar siempre ahí cuando los necesito, por la confianza que siempre tuvieron en mí, por todos los valores que me transmitieron, por forjarme la entereza que tengo al día de hoy y por hacer de mi la persona que en estos momentos soy.
A mis HERMANOS: IVONNE, ROBERTO y CRISTIAN por apoyarme cuando solicite su ayuda y sus ganas de que siempre saliera adelante. Me siento orgullosa de tenerlos como hermanos y sigamos apoyándonos.
A GERARDO, por siempre apoyarme, creer en mí, ayudarme, enseñarme y estar a mi lado en ese
sueño de prepararme y tener una Carrera Profesional que ahora es una realidad. Además de haber pasado tantos momentos en Voca 7 y ESIME Zacatenco.
A mis AMIGOS con quienes compartí mi época estudiantil y todas esas cosas que la Voca 7 y ESIME fueron testigos.
A NELY por apoyarme, por darme su amistad todos estos años, por creer en mí y todos los
momentos que juntas pasamos para terminar la Ingeniería.
A CECI por todo su apoyo.
A mis PROFESORES DE TODAS LAS MATERIAS por transmitirme sus conocimientos, a mis
PROFESORES ASESORES: Ing. Arturo Rojas e Ing. José Antonio Martínez por facilitarme la
elaboración de esta Tesis.
Por último, pero no por eso menos importante al IPN y ESIME por permitirme formar parte de su Comunidad y ser Politécnica de corazón y estar orgullosa de ello y llevar con el mismo orgullo el lema de “La Técnica al Servicio de la Patria”.
GRACIAS por todo y por siempre.
Página i
OBJ ET I VO
Analizar los requisitos del Proyecto de Norma Oficial Mexicana
Página ii
J U ST I FI CACI ÓN
Un aspecto que está tomando gran importancia en la Industria Farmacéutica son sus sistemas automáticos, los cuales requieren cumplir con ciertos requisitos para su validación. Es importante que los estudiantes en la Carrera de Ingeniería en Control y Automatización conozcan el tema ya que esto les permitirá desempeñarse en ese medio, ya sea como Ingenieros de Validación, Ingenieros de Proyectos, Ingenieros de Soporte o Ingenieros de Servicio.
Este conocimiento les permitirá ofrecer sus servicios como Desarrolladores de documentación o Coordinadores / Supervisores de Validación. O en el caso de que formen parte de una empresa de Automatización y Control, que dé servicio y soporte a Laboratorios Farmacéuticos, sean capaces de desarrollar la documentación de diseño y mantenimiento correspondiente a los proyectos que les asignen. Actualmente los Proveedores que dan servicio a la Industria Farmacéutica además de ofrecer un equipo, su instalación y capacitación ofrecen toda la documentación de validación basada en la GAMP4 y es un hecho que no hay Ingenieros con experiencia al respecto.
Si lo vemos desde otra perspectiva este documento puede ser la base para crear una Empresa que ofrezca este Servicio.
Cabe mencionar que nosotros, como Ingenieros en Control y Automatización conocemos la parte técnica de los Sistemas y Procesos, lo que nos facilita desarrollar este tipo de validaciones.
Página iii
I N T RODU CCI ÓN
La Industria Farmacéutica debe cumplir con ciertos requisitos para la fabricación y la distribución de sus productos con el fin de garantizar la Calidad de los mismos. Cada Agencia emite sus leyes o normas para establecer los requisitos que se deben cumplir para la fabricación de los medicamentos que se comercialicen en su País. El control del cumplimiento de estos requisitos en nuestro País es ejercido por la Secretaría de Salud a través de la Norma Oficial Mexicana NOM-059-SSA1-1993.
Dentro de estos requisitos se encuentra la validación de sus Sistemas Computacionales, Equipos Automáticos y la Documentación que esta validación lleva implícita. Los cuáles son los temas que desarrollaré a largo de esta tesis.
Esta validación involucra aspectos como: llevar a cabo el ciclo de vida de los sistemas donde entre otras cosas se definen las especificaciones de requerimientos y de diseño, protocolos de calificación, procedimientos y controles de cambios.
Durante el desarrollo de los temas mencionare términos internacionales usados en inglés ya que son empleados en este contexto.
La lectura de esta tesis le permitirá conocer el proceso de validación de una manera desglosada, práctica, sencilla y con un lenguaje de fácil comprensión. También le dará el punto de partida para profundizar aún más en el tema y adoptar las regulaciones de otros Países.
La tesis se encuentra estructurada en cuatro capítulos, en ellos se destacan los conceptos relevantes que permiten el desarrollo de esta tesis.
En el capítulo primero se describen los antecedentes de la validación de sistemas automáticos y de cómputo desde los años ochentas hasta nuestros días, además del marco normativo nacional e internacional (EU, Australia, Argentina) acerca de este tema.
Página iv
En el capítulo tercero se encuentra la descripción de la propuesta que hago para la Validación de Sistemas Automáticos y de Cómputo en la Industria Farmacéutica. Desde la elaboración del Plan de Validación, el Plan del Proyecto y Calidad, las Especificaciones de Requerimientos y de Diseño, para después la redacción y ejecución de Pruebas llevando la rastreabilidad de los requerimientos hasta llegar a la elaboración del Reporte de Validación. Además de tomar en cuenta el mantenimiento del estado validado del equipo o sistema y los procedimientos de operación. Y por último la elaboración del Plan de Retiro cuando el sistema salga de operación.
El capítulo cuarto describe el Plan de implementación de mi propuesta y los beneficios que aportará.
Página v
Í N DI CE
OBJ ET I V O... i
J U ST I FI CACI ÓN... ii
I N T RODU CCI ÓN... iii
Í N DI CE...v
CAPÍTULO 1. ANTECENTES Y MARCO NORMATIVO...1
1.1 Antecedentes...2
1.2 Marco Normativo ...3
1.2.1 Normatividad Farmacéutica. ... 3
1.2.2 Normatividad Farmacéutica en México. ... 5
CAPÍTULO 2. VALIDACIÓN, CICLO DE VIDA Y GAMP (GOOD AUTOMATED MANUFACTURING PRACTICE, BUENAS PRÁCTICAS DE AUTOMATIZACIÓN PARA MANUFACTURA)...7
2.1 Validación...8
2.1.1 Definición de Validación de Sistemas. ... 8
2.1.2 Introducción al proceso de Validación de Sistemas... 9
2.2 Ciclo de Vida. ...10
2.2.1 Definición de Ciclo de Vida... 10
2.2.2 Elementos de un Ciclo de Vida... 10
2.2.3 Modelos de Ciclo de Vida... 11
2.2.4 Fases del Ciclo de Vida... 14
2.3 La GAMP4 como herramienta para la validación de sistemas automáticos...15
2.3.1 Objetivo de GAMP4 ... 15
2.3.2 Alcance... 15
2.3.3 Visión general de validación en la GAMP4... 15
2.3.4 Actividades y desarrollo de la validación. ... 17
2.3.5 Responsabilidades de usuario. ... 19
2.3.6 Proceso de validación de Sistemas IT... 21
2.3.7 Proceso de validación de Sistemas de Control de Procesos... 23
CAPÍTULO 3. PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA. ...28
3.1 Análisis. ...29
Página vi
3.2 Plan de Validación...37
3.3 Especificación de requerimientos de usuario (URS) y Especificación funcional (FS)..43
3.4 Plan del Proyecto y Calidad (QPP, Quality and Project Plan)...54
3.5 Matriz de Rastreabilidad de Requerimientos...61
3.6 Especificación de Diseño (DS, Desing Specification)...66
3.7 Pruebas al Equipo o Sistema...74
3.7.1 Principios fundamentales que deben seguir las pruebas ... 78
3.7.2 Pruebas de Revisión de Diseño. ... 79
3.7.3 Pruebas de Calificación de Instalación (Installation Qualification, IQ).. 82
3.7.4 Pruebas de Calificación de Operación (Operational Qualification, OQ).83 3.7.5 Pruebas de Calificación de Desempeño (Performance Qualification, PQ)………...………83
3.8 Registro de Capacitación. ...89
3.9 Reporte de Validación. ...89
3.10 Mantenimiento del Estado Validado. ...92
3.11 Retiro. ...97
3.12 Buenas Prácticas. ...102
3.12.1Buenas Prácticas de Documentación... 103
3.12.2Buenas Prácticas de Prueba. ... 103
3.12.3Buenas Prácticas de Ingeniería. ... 105
CAPÍTULO 4. IMPLEMENTACIÓN Y BENEFICIOS...106
4.1 Recursos humanos para la implementación...107
4.2 Inversión para la implementación...108
4.2.1 Costos Directos ... 108
4.3 Compromiso de las Gerencias y de la Dirección...110
4.4 Plan de Implementación. ...111
4.5 Beneficios de la validación...113
4.5.1 Beneficios de negocio. ... 113
4.5.2 Beneficios sociales. ... 117
4.5.3 Beneficios al aplicar mi propuesta ... 117
CONCLUSIONES...119
GLOSARIO Y ACRÓNIMOS ...120
GLOSARIO ...120
ACRÓNIMOS ...123
CAPÍTULO 1. ANTECENTES Y MARCO NORMATIVO
CAPÍTULO 1.
“ANTECEDENTES Y MARCO NORMATIVO”
Objetivo particular:
Describir los antecedentes que dieron lugar al
ANTECEDENTES Y MARCO NORMATIVO
Página 2 de 125
Capítulo 1. Antecedentes y Marco Normativo
Para iniciar con el tema de la Validación de Sistemas Automáticos y de Cómputo empezaré por describir sus antecedentes y la normatividad que regula la Industria Farmacéutica.
1.1 ANTECEDENTES.
En los años ochentas dentro de la normatividad se mencionaba de manera general requerimientos de validación de equipos y software. Pero fue hasta finales de los años ochentas y principios de los noventas donde la validación de sistemas automáticos y de cómputo tomó gran importancia que la que se había tenido hasta ese momento aunque en las regulaciones ya estaban establecidos dichos requerimientos.
Esa importancia fue tomada debido a que a pesar de que las guías concernientes al tema habían estado disponibles ya por algún tiempo, estos sistemas en la Industria Farmacéutica habían sido sometidos a un bajo escrutinio regulatorio en comparación con otras áreas y la interpretación de las guías regulatorias para este ámbito era menos madura que en otras áreas convencionales.
El interés por estos sistemas dentro del ramo farmacéutico se incremento gracias al aumento en su uso y su complejidad, por lo tanto fue necesario mejorar el entendimiento y la interpretación de las regulaciones. Además de una mayor comunicación dentro de la Industria Farmacéutica, así como también con los Proveedores y las Agencias Regulatorias.
Lo anterior aunado a los cambios macroeconómicos y de globalización de esos años como el Acuerdo General sobre Aranceles y Comercio (GATT, General Agreement on Tariffs and Trade), el Tratado de Libre Comercio (TLC) y la creación de la Organización Mundial del Comercio (OMC) han hecho que esto tenga importancia para la importación y exportación de estos productos.
Como dato interesante: entre 1992 y 1998 la FDA (Food and Drug Administration) analizó 3140 retiros del mercado (recalls) de dispositivos médicos, encontrando que 242 de ellos (7.7 %) se atribuían a fallas de software. Dentro de estos 242 casos de software, 192 (79%) fueron causados por defectos de software que se producían por hacer cambios al software después de que la producción y distribución de los productos era iniciada. Estos datos estadísticos refuerzan la importancia en la atención y cumplimiento de los requerimientos en la validación de sistemas automáticos y de cómputo.
Ahora la mayoría de las farmacéuticas están trabajando en cumplir con este requerimiento regulatorio de validación de sistemas automáticos y de cómputo, el cual es solo una parte de toda la regulación que involucra la industria de este ramo.
ANTECEDENTES Y MARCO NORMATIVO
Página 3 de 125 importancia se está dando en el proyecto de Norma PROY-NOM-059-SSA1-2004 donde la sección de validación ya expone los requerimientos que se necesitan para la validación de equipos y sistemas críticos e involucra los sistemas computarizados.
En Australia y Argentina introducen estos requerimientos en 2002 y 2005, respectivamente.
La validación de equipo de cómputo en Estados Unidos es un requerimiento desde 1997 de la regulación de Sistema de Calidad y de CFR 21 (Code of Federal Regulations) Parte 11 correspondiente a Firmas y Registros Electrónicos.
Actualmente existe el Fórum GAMP (Good Automated Manufacturing Practice) que es el grupo que se formó para promover el entendimiento de las regulaciones de equipos automáticos y de cómputo. En nuestros días este Fórum es un subcomité técnico de ISPE (International Society of Pharmaceutical Engineering) y una de sus prioridades, cuando se creó, fue el establecimiento de Guías para Proveedores de sistemas automáticos a la Industria Farmacéutica. La versión preliminar o borrador de estas guías estuvo disponible para comentarios de los Proveedores de dichos sistemas y otros interesados en Marzo 1994. Desde entonces dichas guías se han ido mejorando y actualizando de tal manera que son unas de las Guías más reconocidas a nivel mundial para el cumplimiento de los requerimientos a nivel Internacional. El apego a estas guías facilita el cumplimiento con la FDA y otras Agencias Europeas que son calificadas como las más exigentes en este ámbito.
Ahora abordemos, en la siguiente sección, las características de los requerimientos de la normatividad farmacéutica para ubicarnos en este marco.
1.2 MARCO NORMATIVO
1.2.1 Normatividad Farmacéutica.
Cada País es responsable de emitir su normatividad en cuestión de fabricación de medicamentos con el fin de que se garantice la calidad de los mismos para su población.
En México le corresponde a la Secretaría de Salud establecer los requerimientos para tal fin y lo hace empleando como marco de referencia la Norma Oficial Mexicana NOM-059-SSA1-1993, Buenas Prácticas de fabricación para establecimientos de la industria químico farmacéutica dedicados a la fabricación de medicamentos. Las secciones 5.6.3, 5.7.4 y 9.11 especifican algunos aspectos de validación. Sin embargo se tiene el Proyecto de Norma Oficial PROY-NOM-059-SSA1-2004, donde se detallan un poco más estos requerimientos en las secciones 10.5 y 14. En las cuales se establecen los requerimientos para los Equipos automáticos, mecánicos y electrónicos y para la Validación.
ANTECEDENTES Y MARCO NORMATIVO
Página 4 de 125 FDA (Food and Drug Administration). Las secciones que corresponden a las Buenas Prácticas de Manufactura para productos farmacéuticos son Parte 210 y 211, la de firmas y registros electrónicos es la Parte 11 y la de Validación de Software y equipos automáticos se encuentra dentro de la Parte 820.
Entre otros países, por ejemplo, podemos citar Australia con la TGA (Therapeutic Goods Administration) y su código Australian Code of Good Manufacturing Practice for Medicinal Products. Donde en el Anexo 11 menciona los requerimientos a cumplir para los sistemas computarizados y en el Anexo 15 los referentes a la Calificación y Validación.
Y Argentina con la ANMAT (Administración Nacional de Medicamentos, Alimentos y Tecnología Médica) por medio de la Disposición 2819/2004, especifica los requerimientos para la Calificación y Validación en la sección 4 y en el Anexo II.
La siguiente lista presenta algunos de los requerimientos que son solicitados en las regulaciones de México, EU, Argentina y Australia.
Elaboración de un Plan de Validación.
Capacitación de Personal.
La validación debe ser considerada como parte del ciclo de vida de unsistema computarizado.
Especificaciones de Diseño.
El software es un componente crítico, que debe ser producido conformeun sistema de Aseguramiento de Calidad.
Seguridad para el acceso a los sistemas (control de acceso y rastreo de auditoría).
Control de cambios.
Respaldos, resguardo y recuperación.
Rastreabilidad.
Atención de fallas y problemas.
Calificación de Diseño.
Calificación de Instalación.
Calificación de Operación.
Calificación de Desempeño.
Mantenimiento del estado validado.
Control de cambios.
Revalidación.
Auditoría de Rastreo.
Firmas y registros electrónicos.ANTECEDENTES Y MARCO NORMATIVO
Página 5 de 125
1.2.2 Normatividad Farmacéutica en México.
Dentro de la NOM-059-SSA1-1993 indica que se deben llevar a cabo estudios de validación para los sistemas involucrados en los procesos de fabricación. Y con respecto a los equipos automáticos y electrónicos señala que deben ser calibrados, inspeccionados y asegurar y respaldar la información que manejan. En esta versión vigente de la Norma se puede observar que no se desglosan los requerimientos necesarios para cumplir con la validación señalada, pero actualmente existe el Proyecto de Norma Oficial PROY-NOM-059-SSA1-2004 en el cual ya se toman en consideración estos puntos.
El PROY-NOM-059-SSA-2004 específica que los equipos, sistemas críticos y computacionales que impacten la calidad del producto deben ser calificados y validados. Para llevar a cabo esta validación nos presenta los requerimientos que la integran, a grandes rasgos son los siguientes.
Hacer un Plan de la validación
Documentación
Calificación
Llevar un control de cambios en este sentido
Mantener el Sistema ValidadoEs un hecho que si se comparan los requerimientos que marca el Proyecto de Norma Mexicana para los sistemas computacionales con los que solicitan las Normas de Australia, Argentina y/o EU, que estoy tomando como referencia, resultan muy básicos y todavía ambiguos.
La sección del proyecto de norma mexicana que hace referencia a los sistemas computacionales es la 14.8 y solo hace referencia a lo siguiente:
14.8 Sistemas computacionales.
14.8.1 Deben validarse los sistemas y aplicaciones computacionales relacionados con:
14.8.1.1 Transferencias de materiales y producto.
14.8.1.2 Disposición de materiales y producto.
14.8.1.3 Control de procesos y análisis.
14.8.1.4 Control de sistemas críticos.
Y si lo comparamos por ejemplo con la de Australia que tiene todo un anexo dedicado al punto de sistemas computarizados donde tiene 22 puntos con los requerimientos respecto al tema vemos que el proyecto de norma sigue un poco deficiente con el cumplimiento a nivel internacional.
ANTECEDENTES Y MARCO NORMATIVO
Página 6 de 125 muchos puntos pendientes por cubrir para alcanzar los requerimientos de otras legislaciones.
Por ejemplo el PROY-NOM-059-SSA-2004 no contempla requerimientos acerca de auditoría de rastreo (audit trail) para los sistemas computarizados, los cuales si son mencionados por la TGA y FDA.
Además hasta este PROY-NOM-059-SSA-2004 se solicita la existencia de un Plan para definir los requerimientos del producto, procesos, sistemas críticos y servicios y que su diseño esté conforme a los requerimientos definidos. Este punto es muy importante ya que está directamente relacionado con la ISO 9001:2000 en la sección de Diseño.
CAPÍTULO 2. VALIDACIÓN, CICLO DE VIDA Y GAMP
(GOOD AUTOMATED MANUFACTURING PRACTICE,
BUENAS PRÁCTICAS DE AUTOMATIZACIÓN PARA
MANUFACTURA)
CAPÍTULO 2.
“VALIDACIÓN, CICLO DE VIDA Y GAMP
(GOOD AUTOMATED MANUFACTURING
PRACTICE, BUENAS PRÁCTICAS DE
AUTOMATIZACIÓN PARA MANUFACTURA).”
Objetivos particulares:
Introducir al proceso de validación de los sistemas automáticos
y de cómputo.
Describir el Ciclo de Vida.
VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 8 de 125
Capítulo 2. Validación, Ciclo de Vida y GAMP (Good
Automated Manufacturing Practice, Buenas Prácticas de
Automatización para Manufactura).
Después de conocer los antecedentes generales y el marco normativo de la validación de sistemas computarizados, abordemos la definición de la Validación, el tema del Ciclo de Vida y los objetivos de la Guía de las Buenas Prácticas de Automatización para Manufactura.
2.1 VALIDACIÓN.
2.1.1 Definición de Validación de Sistemas.
Hablar de validación comprende varios aspectos del proceso de manufactura farmacéutica desde los procesos, sistemas, equipos y servicios hasta la limpieza de las áreas. Pero empecemos por ver que hay alrededor de la palabra Validación.
A continuación se muestran algunas definiciones del Proceso de Validación.
La NOM-059-SSA1-1993 nos indica que la validación es “la evidencia documentada que demuestra que a través de un proceso específico se obtiene un producto que cumple consistentemente con las especificaciones y los atributos de calidad establecidos”.
Por otro lado la definición de Validación que nos dan en la GAMP4 es
“establishing documented evidence which provides a high degree of assurance that a specific process will consistently produce a product meeting its pre-determined specifications and quality attributes”. En español “establecer evidencia documentada que provea un alto grado de aseguramiento de que un proceso específico producirá consistentemente un producto cumpliendo con especificaciones y atributos de calidad predeterminados”.
En el Código Australiano de Buenas Prácticas de Manufactura para Productos
Medicinales podemos encontrar que el Proceso de Validación es “the documented
VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 9 de 125 De las definiciones anteriores se puede rescatar las siguientes ideas:
La validación es una evidencia documentada de un proceso.
A través de ese proceso se obtiene un producto, que en este caso es unproducto farmacéutico.
Este proceso y producto cumplen con especificaciones y atributos decalidad predeterminados.
El proceso de fabricación de ese producto debe ser eficaz y reproducible.Por lo tanto, reuniendo las ideas anteriores, la validación es la evidencia documentada de que a través de un proceso eficaz y reproducible, un producto farmacéutico es obtenido de acuerdo a las especificaciones, atributos de Calidad predeterminados y principios de buenas prácticas de manufactura.
La definición está de alguna manera apegada al proceso de obtención un producto, pero sin embargo si hablamos de un sistema de manera general podemos decir que la validación de sistemas es la generación de evidencia documentada que nos proporcione un alto grado de aseguramiento de que el sistema funcionará como se requiere cuando se ponga en uso.
2.1.2 Introducción al proceso de Validación de Sistemas.
La importancia de validar un sistema es que nos asegura su funcionamiento y el beneficio que se obtiene es el Cumplimiento Legal. La validación involucra que los sistemas críticos y los equipos de producción deben ser probados tomando en cuenta su diseño, su instalación y su operación.
Cuando se tiene un sistema nuevo por instalar, su validación será de una manera gradual y tomando en cuenta algo muy importante: la participación y compromiso del usuario y del proveedor.
En otras palabras validar un sistema involucra:
Tener su Plan de validación y aseguramiento de la Calidad.
Desarrollo de documentos donde se especifique los requerimientos y eldiseño del sistema.
Desarrollo de Planes de Prueba y Protocolos de Calificación de Instalación, Operación y Desempeño.
Sus cambios deben estar controlados.
Desarrollo de PNO’s para su administración.
Ser un sistema seguro, es decir, que tenga protección que no permita lamodificación o perdida de datos.
Tener respaldo de la información.VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 10 de 125
Mantenimiento y seguimiento del estado validado.
Plan de Retiro.2.2 CICLO DE VIDA.
En algunos casos se hace referencia al termino Ciclo de Vida del Sistema y en otros al termino Ciclo de Vida del proyecto, en realidad cualquiera de los dos términos son correctos depende del contexto en donde se emplee sólo habrá que ver si en realidad se tiene un proyecto como tal o si se está validando a un sistema que no pertenece a un proyecto pero en ambos se está utilizando el concepto de Ciclo de Vida como herramienta para su desarrollo.
2.2.1 Definición de Ciclo de Vida.
El Ciclo de Vida se define como un conjunto de fases sucesivas empleadas para
generar un producto, un proceso o un servicio. Cada una de estas fases sucesivas se divide a su vez en actividades específicas para el cumplimiento del objetivo de cada fase. El agrupamiento de estas actividades depende, además del objetivo de cada fase, del producto, proceso o servicio final que se pretende obtener y de los requerimientos que haya que cumplir para su obtención.
El dividir los proyectos o los procesos en fases nos asegura la reducción de la complejidad de los mismos. El empleo del concepto de Ciclo de Vida nos permite:
Definir las actividades a ser ejecutadas.
Tener un mayor control sobre los tiempos.
Subcontratar servicios (para el desarrollo de actividades específicas).
Controlar el logro del objetivo principal mediante la obtención de objetivos específicos.
Tomar decisiones al término de cada fase en base a los resultadosobtenidos.
2.2.2 Elementos de un Ciclo de Vida.
Los elementos que integran un Ciclo de Vida son:1
Fase (s): Una fase dentro del Ciclo de Vida es un conjunto de actividades relacionadas entre sí con un objetivo dentro del desarrollo de un proyecto o proceso.El detalle en la definición de cada fase dependerá del tamaño del proyecto y de su complejidad. El propósito de la división en fases es que el contenido de cada una sea manejable, controlado y con una relación lo más simple posible con las demás fases.
1
VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 11 de 125
Entregables: Son los productos intermedios que se deben generar en cada fase. En inglés el término utilizado para los entregables es“deliverables”.
Estos productos intermedios pueden ser equipos, componentes, documentos, programas, software, manuales, entre otros. Los entregables al final de cada fase del proyecto nos ayudan a evaluar la marcha del proyecto en cumplimiento con los requisitos previamente establecidos.
2.2.3 Modelos de Ciclo de Vida.
Los modelos de Ciclo de Vida son:1
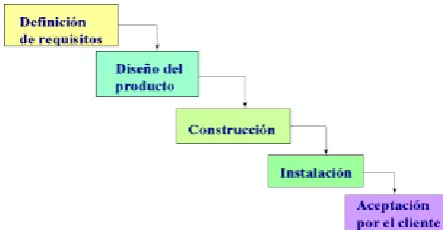
Ciclo de Vida Lineal.Este modelo consiste en descomponer el proyecto o el proceso en fases que suceden de manera lineal. Cada fase se realiza una sola vez y siempre tienen una secuencia, cada fase tiene una fase o actividad precedente y una fase o actividad que le procede. Este modelo consta de un solo ciclo.
[image:19.595.202.424.469.584.2]Una de las características de este modelo es que el proyecto o proceso se pueda dividir de manera que cada fase no necesite realimentación de otra, es decir, que el resultado obtenido de una fase no realimente a una fase anterior. Aunque es valida la realimentación correctiva. Este modelo es el más empleado por su sencillez. Ver figura 2.1.
Figura 2.1. Ejemplo de Modelo de Ciclo de Vida Lineal.
1
VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 12 de 125
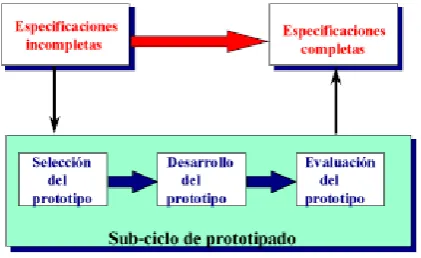
Ciclo de Vida con Prototipado. [image:20.595.210.421.252.381.2]Este modelo es empleado cuando no se conocen con exactitud las especificaciones de un producto o proyecto y es necesario tener una retroalimentación de los resultados obtenidos, evaluarlos, hacer correcciones y entonces obtener las especificaciones completas. En este modelo regularmente se definen especificaciones iniciales para hacer un prototipo (un producto parcial y provisional), se desarrolla y en base a los resultados se repiten las fases de definición, diseño y construcción; con lo que llevan a cabo dos ciclos. Comúnmente este modelo es empleado en desarrollo avanzado. Ver figura 2.2.
Figura 2.2 Ejemplo de Modelo de Ciclo de Vida Prototipado.
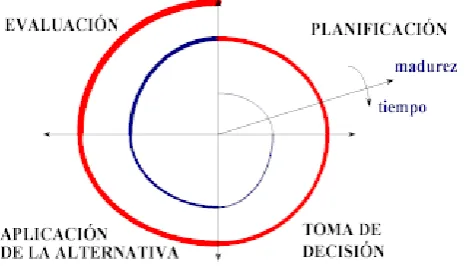
Ciclo de Vida en espiral.Este modelo es empleado cuando no basta con una sola evaluación del prototipo, como en el modelo anterior, de modo que es necesario hacer más evaluaciones y correcciones para eliminar incertidumbres en las especificaciones y diseño del sistema, producto o proyecto. En este modelo se tienen más de dos ciclos, y en cada uno de ellos se obtiene un prototipo mejorado hasta alcanzar el prototipo deseado, ya que en cada ciclo se van resolviendo las dudas de las especificaciones.
VALIDACIÓN, CICLO DE VIDA Y GAMP
[image:21.595.200.433.106.239.2]Página 13 de 125
Figura 2.3. Ejemplo de Modelo de Ciclo de Vida en Espiral.
VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 14 de 125
2.2.4 Fases del Ciclo de Vida.1
El ciclo de vida se clasifica en fases o etapas, a continuación muestro las que nos sugieren en el artículo Ciclo de Vida en la página de Internet citada al pie de esta página.
Fase de definición (¿qué hacer?)
Estudio de viabilidad.
Conocer los requisitos que debe satisfacer el sistema (funciones y limitaciones de contexto).
Asegurar que los requisitos son alcanzables.
Formalizar el acuerdo con los usuarios.
Realizar una planificación detallada.Fase de diseño (¿cómo hacerlo? Soluciones en costo, tiempo y calidad)
Identificar soluciones tecnológicas para cada una de las funciones del sistema.
Asignar recursos materiales para cada una de las funciones.
Proponer (identificar y seleccionar) subcontratar.
Establecer métodos de validación del diseño.
Ajustar las especificaciones del producto.Fase de construcción
Generar el producto o servicio pretendido con el proyecto.
Integrar los elementos subcontratados o adquiridos externamente.
Validar que el producto obtenido satisface los requisitos de diseñopreviamente definidos y realizar, si es necesario, los ajustes necesarios en dicho diseño para corregir posibles lagunas, errores o inconsistencias.
Fase de mantenimiento y operación
Operación: asegurar que el uso del proyecto es el pretendido.
Mantenimiento (nos referimos a un mantenimiento no habitual, es decir, aquel que no se limita a reparar averías o desgastes habituales -este es el caso del mantenimiento en productos software, ya que en un programa no cabe hablar de averías o de desgaste):
1
VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 15 de 125
2.3 LA GAMP4 COMO HERRAMIENTA PARA LA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS.
Dentro del ámbito farmacéutico se ha desarrollado una herramienta que proporciona una ayuda para la validación y el cumplimiento de las expectativas regulatorias actuales para los sistemas automáticos, es decir, es una guía para validar dichos
sistemas. Esta guía es llamada GAMP por sus siglas en Inglés Good Automated
Manufacturing Practice, en español podemos decir que son las Buenas Prácticas de Automatización para Manufactura.
Esta guía surgió a partir de la necesidad de la interpretación y entendimiento de las regulaciones, debido a que las reglas estaban establecidas pero se requería una mayor comunicación e uniformidad en los conceptos.
Por otro lado también proporciona una guía para el desarrollo y mantenimiento de las Buenas Prácticas, ya que en su conjunto cubre todos los requerimientos para la validación y cumplimiento con las Buenas Prácticas de Manufactura.
Una de las características de la GAMP4 es que no está dirigida a un solo grupo de personas. Está dirigida a usuarios y proveedores de sistemas automáticos, ambos de la Industria Farmacéutica.
2.3.1 Objetivo de GAMP4
Su objetivo es asistir a las Industrias que se dedican al cuidado de la Salud, incluyendo Farmacéutica, Biotecnológica y de Dispositivos Médicos, a lograr la validación y cumplimiento de los sistemas automáticos.
2.3.2 Alcance
La GAMP4 proporciona un alcance general para todos los sistemas automáticos,
aunque reconoce que todos los sistemas no son iguales y tienen diferencias significantes.
Esta guía aplica para desarrollar el Ciclo de Vida de sistemas nuevos. Los sistemas existentes no entran en el alcance de esta guía.
Por otro lado la guía especifica que no todas las actividades descritas aplican a todos los proyectos, ya que el líder de proyecto y/o el área usuaria deberán de especificar los requerimientos de validación necesarios para cada sistema.
2.3.3 Visión general de validación en la GAMP4
VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 16 de 125
IQ Calificación de la Instalación por sus siglas en Inglés Installation Qualification.
OQ Calificación de Operación por sus siglas en Inglés OperationalQualification.
PQ Calificación de Desempeño por sus siglas en Inglés PerformanceQualification.
DQ Calificación de Diseño por sus siglas en Inglés Design Qualification.Y nos presentan un esquema general de las actividades de validación que se muestra a continuación:
Especificación y acuerdos de lo que se requiere. Elaborar las revisiones de Diseño.
PLANEACIÓN
ESPECIFICACIÓN
PLAN DE PRUEBAS (IQ, OQ,PQ)
PRUEBAS (IQ, OQ,PQ)
REVISIÓN DE RESULTADOS
La Planeación se llevará a cabo mediante la elaboración del Plan de Validación.
Elaboración de un documento donde se describa como se probará el sistema.
Ejecución de pruebas y colección de resultados.
[image:24.595.85.510.256.589.2]Conclusión de resultados para mostrar que el sistema funciona como fue especificado.
Figura 2.4 Actividades Generales de Validación.
VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 17 de 125
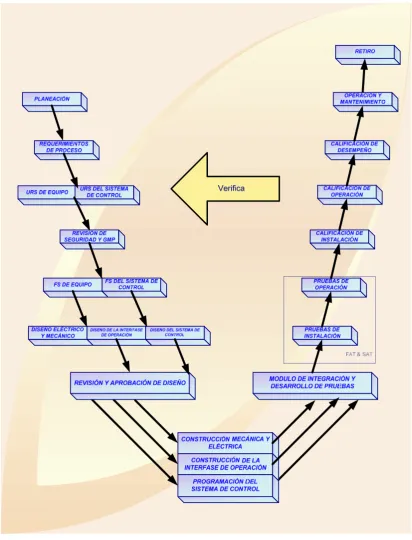
[image:25.595.120.478.103.479.2]Verifica
Figura 2.5 Estructura básica y relación de Especificaciones y Pruebas de Calificación.
2.3.4 Actividades y desarrollo de la validación.
VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 18 de 125 REVISIONES DE
CÓDIGO
Actividades a desarrollar
Fases en el Ciclo de Vida
Actividades de Validación
Especificación de Requerimientos de Usuario Especificaciones Funcionales
Especificación de Diseño de Hardware Especificación de Diseño de Software Especificación de Diseño del Modulo de Software Especificaciones Mecánicas y Eléctricas Especificación de Diseño de Red
Especificación de Configuración del Paquete o Empaque
Manufactura y Ensamble del Hardware Módulos de Código de Software Manufactura y Ensamble de Equipo Manufactura y Ensamble de Red
Pruebas de hardware Pruebas del Módulo de Software Pruebas del Integración de Software Pruebas de equipo
Pruebas de la Configuración del Paquete o Empaque
Instalación de Hardware Instalación de Software Instalación de Equipo Instalación de Red
Pruebas de aceptación de hardware Pruebas de aceptación de red
Pruebas de aceptación de Sistema
Mantenimiento Control de Cambios
PLANEACIÓN Y ESPECIFICACIÓN DISEÑO CONSTRUCCIÓN PRUEBA INSTALACIÓN
PRUEBAS DE INSTALACIÓN
[image:26.595.85.512.114.662.2]OPERACIÓN PLAN DE VALIDACIÓN EVALUACIÓN DEL PROVEEDOR REVISIONES DE DISEÑO REVISIONES DE DISEÑO REVISIONES DE CONSTRUCCIÓN MONITOREO DEL PROVEEDOR CALIFICACIÓN DE INSTALACIÓN CALIFICACIÓN OPERACIONAL CALIFICACIÓN DE DESEMPEÑO REPORTE DE VALIDACIÓN MANTENIMIENTO DEL ESTADO VALIDADO
VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 19 de 125 La figura anterior 2.6 muestra de manera general las actividades de validación, pero por otro lado, la GAMP4 para abordar el proceso de validación hace una diferencia entre los sistemas de Tecnología de la Información (IT) y los Sistemas de Control de Proceso. Para el propósito de dicha guía un Sistema de Tecnología de la Información (IT) incluye Planificación de Recursos de la Empresa (ERP, Enterprise Resource Planning), Sistemas de Administración de Información de Laboratorio (LIMS, Laboratory Information Management System), administración de producción y sistemas de base de datos. Y un sistema de control de proceso incluye grandes sistemas de control como los de Adquisición de Datos y Supervisión de Control (SCADA, Supervisory Control and Data Acquisition), Sistemas de Control Distribuido (DCS, Distributed Control System), sistemas controlados por un Controlador Lógico Programable (PLC, Programmable Logic Controller) y sistemas integrados (embedded systems) instalados en manufactura.
A continuación les presento las responsabilidades del usuario en el proceso de validación, el proceso de validación para los sistemas IT y para los Sistemas de Control de Proceso que nos sugiere la GAMP4.
2.3.5 Responsabilidades de usuario.
La GAMP4 en su capítulo de “Ciclo de Vida de la Validación” (“Validation Life Cycle”) nos explica las responsabilidades que tiene el usuario dentro del proceso de
VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 20 de 125
No. DE
PASO TAREA O ACTIVIDAD DESCRIPCIÓN
1 Identificación del Sistema. Cada sistema automático debe ser evaluado e identificado como Sistema regulatorio GMP.
2 Elaboración del URS.
El URS debe definir de manera clara y precisa lo que el usuario quiere que haga el sistema, algunas restricciones y definir requerimientos regulatorios y de documentación.
3
Determinar la estrategia de validación * Evaluación del riesgo.
* Estimación de los componentes del sistema. * Evaluación del proveedor.
Una evaluación de riesgo inicial debe ser llevada durante el Plan de Validación.
Los componentes del sistema deben ser estimados y categorizados para determinar el acercamiento a la validación requerida.
Los proveedores deben ser formalmente evaluados como parte del proceso de selección a proveedores y la planeación para la validación. La decisión de si se realizará una auditoría a Proveedores debe ser documentada y basada en la evaluación de riesgo y en la categorización de componentes.
4 Elaboración del Plan de Validación.
El Plan de Validación debe definir las actividades, procedimientos y responsabilidades para establecer el sistema. Es típico definir la evaluación de riesgo que será tomada.
5 Revisión y aprobación de especificaciones, incluyendo la descripción del sistema. El usuario debe revisar y aprobar las especificaciones elaboradas por el proveedor.
6 Monitoreo del desarrollo del Sistema. El usuario debe monitorear el desarrollo y las actividades de configuración contra el Plan acordado.
7 Revisión del código fuente. El usuario debe asegurar que el código fuente sea revisado adecuadamente durante el desarrollo del Sistema.
8 Revisión y aprobación de las especificaciones de prueba.
El usuario debe revisar y aprobar las especificaciones de prueba antes de ejecutarlas formalmente.
9 Desarrollo de las pruebas. El usuario puede ser involucrado en las pruebas como testigo durante la ejecución o como revisor de los resultados.
10 Revisión y aprobación de los reportes de prueba. El usuario debe aprobar los reportes de prueba y los resultados asociados.
11 Elaboración del Reporte de Validación.
El reporte de validación debe resumir todos los entregables y actividades, además de proveer evidencia de que el sistema está validado.
12 Mantenimiento del Sistema.
Una vez que el sistema ha sido aceptado, el usuario debe establecer los procedimientos adecuados para la administración y operación del sistema.
[image:28.595.84.511.118.672.2]13 Retiro del Sistema. El usuario debe administrar el reemplazo o el retiro del uso del sistema automático.
VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 21 de 125
2.3.6 Proceso de validación de Sistemas IT.
La GAMP4 aborda el proceso de validación de los sistemas IT a través de los proveedores. En su capítulo “Sistema de Administración para los proveedores de Sistemas de Tecnología de la Información” (“Management System for Suppliers of IT Systems”) nos sugiere un modelo de validación basado en el concepto de ciclo de
vida.
Con el objetivo de que los proveedores jueguen su rol en la producción de sistemas validados la GAMP4 recomienda que los proveedores sigan un sistema de administración formal y de preferencia basado en estándares tales como las series de la ISO 9000. Esta sección de la Guía sugiere un modelo basado en el concepto de ciclo de vida para el desarrollo y soporte subsecuente de un sistema automático.
El modelo descrito va encaminado a proveedores de Sistemas de Tecnología de la Información, como ERP, LIMS, administración de la producción y sistemas de base de datos.
Muchos sistemas IT para la industria farmacéutica actualmente están basados en productos de software y paquetes, los cuales están configurados cumpliendo los requerimientos del usuario. De tal manera que estos productos vienen con documentación de soporte y donde sea posible esta información puede ser empleada en la validación del ciclo de vida. Adicionalmente módulos de software son a veces requeridos para ofrecer o satisfacer una funcionalidad específica tal como interfases y reportes. El diseño y desarrollo de cada software debe ser documentado.
Esta sección aplica tanto para sistemas configurados específicamente como para sistemas de solución común o para la combinación de ambos.
El modelo de ciclo de vida que nos sugiere la Guía para validar estos sistemas esta mostrado en la figura 2.7. Este modelo cubre las fases de Planeación, Especificación, Diseño, Construcción, Pruebas, Instalación, Pruebas de aceptación y Operación.
VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 22 de 125
(Mantenimiento del estado validado)
PLANEACIÓN Y ESPECIFICACIÓN
ESPECIFICACIÓN DE REQUERIMIENTOS DE
USUARIO PLAN DE VALIDACIÓN
GAMP4
EVALUACIÓN DEL PROVEEDOR
PLAN DEL PROYECTO Y CALIDAD ESPECIFICACIÓN FUNCIONAL DISEÑO Y CONSTRUCCIÓN INSTALACIÓN Y PRUEBAS
PRUEBAS DE ACEPTACIÓN
OPERACIÓN
ESPECIFICACIÓN DE DISEÑO DEL
HARDWARE ESPECIFICACIÓN DE PRUEBAS DE ACEPTACIÓN DEL HARDWARE ESPECIFICACIÓN DE DISEÑO DEL
SOFTWARE ESPECIFICACIÓN DE PRUEBAS DE INTEGRACIÓN DEL SOFTWARE ESPECIFICACIÓN DE CONFIGURACIÓN DEL PAQUETE ESPECIFICACIÓN DE PRUEBAS DE CONFIGURACIÓN DEL PAQUETE ESPECIFICACIÓN DE DISEÑO DEL MODULO
DE SOFTWARE
ESPECIFICACIÓN DE PRUEBAS DEL MODULO DE
SOFTWARE
PRODUCCIÓN DE
HARDWARE PRODUCCIÓN DE SOFTWARE CONFIGURACIÓNPAQUETE DE
RESULTADOS Y PRUEBAS DE ACEPTACIÓN DE HARDWARE RESULTADOS Y PRUEBAS DEL MODULO DE SOFTWARE RESULTADOS Y PRUEBAS DE INTEGRACIÓN DE SOFTWARE
[image:30.595.83.513.117.670.2]VERIFICACIÓN DE LA CONFIGURACIÓN RESULTADOS Y PRUEBAS DE ACEPTACIÓN DEL SISTEMA MANTENIMIENTO CONTROL DE CAMBIOS ACEPTACIÓN DEL SISTEMA ESPECIFICACIONES DE PRUEBA
VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 23 de 125
2.3.7 Proceso de validación de Sistemas de Control de Procesos.
La GAMP4 en su capitulo “Validación de Sistemas de Control de Procesos”
(“Process Control System Validation”) considera cómo las actividades de ciclo de
vida pueden ser aplicadas a un rango de control, monitoreo, o sistemas analíticos diseñados a administrar la producción o procesos de laboratorio.
Los sistemas de control de proceso son usados para automatizar los procesos de manufactura o de laboratorio por medio de colección de datos que provienen de instrumentos de medición o a través de algoritmos de control o bloques lógicos pre-programados o configurados, regulando dispositivos finales de control como válvulas o bombas para controlar equipos y/o procesos.
La función primaria de un sistema de control de proceso es producir un producto o proveer datos para soporte de manufactura o de laboratorio. Algunos sistemas de control de proceso también proveen funcionalidad en la administración de los procesos o están ligados con el procesamiento de datos de aplicaciones como parte de una computadora integrada a manufactura.
Las buenas prácticas de ingeniería deben ser aplicadas al desarrollo e implementación de sistemas de control de proceso. Las buenas prácticas de ingeniería son la aplicación de los métodos o estándares establecidos a través del proyecto de ciclo de vida para entregar apropiadamente soluciones costo-beneficio.
2.3.7.1 Modelos de Ciclo de Vida
La validación del ciclo de vida de un Sistema de Control de Proceso está definida en la figura 2.8 y aplica para un amplio rango de sistemas, desde sistemas de control robusto en industrias grandes hasta sistemas de control pequeños instalados en equipos usados como secundarios o ligados con equipo de laboratorio montado.
La relación entre las fases de diseño y de pruebas es compleja. La verificación de que se ha conocido la intención de los requerimientos y diseño debe hacerse en la parte mas apropiada del ciclo de vida dependiendo del tipo y madurez del sistema que está siendo desarrollado o configurado, del nivel del plan de pruebas y de la complejidad de los requerimientos a probar.
VALIDACIÓN, CICLO DE VIDA Y GAMP
[image:32.595.91.504.157.698.2]Página 24 de 125 La figura 2.8 ilustra el ciclo de vida para la validación de un sistema de control de proceso con su consecutivo proceso de control y niveles de especificación de diseño y sus revisiones y calificaciones correspondientes.
VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 25 de 125 2.3.7.2 Tipos de Sistema de Control de Proceso
La GAMP4 distingue dos Sistemas de Control de Proceso: Sistemas Integrados (Embedded Systems) y Sistemas Autónomos (Standalone Systems).
Sistemas Integrados (Embedded Systems): Son Sistemas basados en microprocesadores tales como un Circuito Integrado Programable (IC), un Controlador Lógico Programable (PLC) o una Computadora con el solo propósito de controlar o monitorear una pieza o parte de manufactura o equipo analítico, el cual es usualmente entregado como una parte de ese determinado equipo. Ejemplos: máquinas de llenado, máquinas de empaque o Cromatógrafos de Líquidos de Alto Desempeño (HPLC, High performance liquid chromatography).
En la figura 2.9 podemos ver el modelo que nos presenta la GAMP4 para este tipo de sistemas.
Sistemas Autónomos (Standalone Systems): Son aquellos sistemas que son configurados o hechos a la medida, sistemas contenidos en si mismos que son componentes de una aplicación de un proceso automático de manufactura. Éstos son integrados como sistemas de cómputo independientes, separados del equipo de Planta, para su conexión a instrumentación de campo o dispositivos reguladores y a cada uno. Estos sistemas pueden ser integrados verticalmente con un nivel alto de administración de datos. Ejemplos: controladores multi-loop o Controlador
Lógico Programable (PLC, Programmable Logic Controller) que controlan parte
de un proceso, sistemas de Adquisición de Datos y Supervisión de Control (SCADA, Supervisory Control and Data Acquisition), Sistemas de Control Distribuido (DCS) que controlen procesos de Planta y Sistemas de Administración de Edificios (BMS, Building Management Systems).
VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 26 de 125 RESPONSABILIDAD CONTROL DE CAMBIOS ESPECIFICACIÓN DE PRUEBAS DE SOFTWARE PRUEBAS DE SOFTWARE Y RESULTADOS INSTALACIÓN Y LIBERACIÓN DE SOFTWARE
ESPECIFICACIÓN DE PRUEBAS DE ACEPTACIÓN DE FABRICA CONSTRUCCIÓN MECÁNICA CABLEADO PLAN DE VALIDACIÓN ESPECIFICACIÓN DE REQUERIMIENTOS DEL USUARIO EVALUACIÓN DEL PROVEEDOR PARÁMETROS CRÍTICOS DE PROCESO APROBACIÓN DE LA ESPECIFICACIÓN EVALUACIÓN DEL PROVEEDOR ELÉCTRICA E INSTRUMENTACIÓN SOFTWARE MECÁNICA ESPECIFICACIÓN DE DISEÑO DIAGRAMAS ELÉCTRICOS & DE INSTRUMENTACIÓN Y
HARDWARE
ESPECIFICACIÓN DE DISEÑO DE SOFTWARE
CÓDIGO DIAGRAMAS
MECÁNICOS
PRUEBAS Y RESULTADOS DE ACEPTACIÓN DE FABRICA
PRUEBAS ELÉCTRICAS & CALIBRACIÓN DE INSTRUMENTOS Y RESULTADOS ESPECIFICACIÓN DE PRUEBAS DE ACEPTACIÓN
DE HARDWARE CALIFICACIÓN DE INSTALACIÓN CALIFICACIÓN OPERACIONAL CALIFICACIÓN DE DESEMPEÑO GEP: INSTALACIÓN PRUEBAS DE ACEPTACIÓN DE SITIO COMMISSIONING
MANTENIMIENTO PERIÓDICAREVISIÓN
RETIRO UBICACIÓN CLIENTE PROVEEDOR PROVEEDOR PROVEEDOR PROVEEDOR CLIENTE CLIENTE CLIENTE CLIENTE CLIENTE CLIENTE CLIENTE (PROVEEDOR) PROVEEDOR (CLIENTE) PROVEEDOR (CLIENTE) CLIENTE (PROVEEDOR) CLIENTE (PROVEEDOR) FAT: PROVEEDOR (CLIENTE) REVISIÓN DE DISEÑO: CLIENTE (PROVEEDOR) INSTALACIÓN: PROVEEDOR (CLIENTE) CLIENTE (PROVEEDOR) IQ: SAT: PROVEEDOR (CLIENTE) CLIENTE (PROVEEDOR) OQ: COMMISSIONING: PROVEEDOR (CLIENTE) CLIENTE (PROVEEDOR) PQ: PLANEACIÓN Y DEFINICIÓN ESPECIFICACIÓN DE DISEÑO, DESARROLLO Y CONSTRUCCIÓN
REVISIÓN DE DISEÑO Y PRUEBAS COMMISSIONING Y CALIFICACIÓN OPERACIÓN CONTINUA (MANTENIMIENTO DEL ESTADO VALIDADO) DE-COMMISSIONING / RETIRO
NOTA: EL RESPONSABLE ENTRE PARÉNTESIS INDICA
[image:34.595.84.531.105.686.2]POSIBLE PARTICIPACIÓN
VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 27 de 125 RESPONSABILIDAD
PLAN DE VALIDACIÓN
ESPECIFICACIÓN DE REQUERIMIENTOS DEL USUARIO EVALUACIÓN DEL PROVEEDOR PARÁMETROS CRÍTICOS DE PROCESO
APROBACIÓN DE LA ESPECIFICACIÓN EVALUACIÓN DEL PROVEEDOR UBICACIÓN CLIENTE PROVEEDOR CLIENTE CLIENTE (PROVEEDOR) PLANEACIÓN Y DEFINICIÓN
NOTA: EL RESPONSABLE ENTRE PARÉNTESIS INDICA
POSIBLE PARTICIPACIÓN CLIENTE CLIENTE CLIENTE CLIENTE PRUEBAS DE ACEPTACIÓN DE FABRICA
(CONTRA PUNTO 1)
CONTROL DE CAMBIOS DATOS DE DISEÑO DE PROCESO CALIFICACIÓN DE INSTALACIÓN CALIFICACIÓN OPERACIONAL CALIFICACIÓN DE DESEMPEÑO GEP: INSTALACIÓN PRUEBAS DE ACEPTACIÓN DE SITIO
COMMISSIONING
MANTENIMIENTO REVISIÓN PERIÓDICA
RETIRO
R
EVIS
IÓN DEL DIS
E ÑO PROVEEDOR PROVEEDOR PROVEEDOR CLIENTE CLIENTE CLIENTE CLIENTE PROVEEDOR (CLIENTE) PROVEEDOR (CLIENTE) CLIENTE (PROVEEDOR) CLIENTE (PROVEEDOR) FAT: PROVEEDOR (CLIENTE) REVISIÓN DE DISEÑO:
CLIENTE (PROVEEDOR) INSTALACIÓN: PROVEEDOR (CLIENTE) CLIENTE (PROVEEDOR) IQ: SAT: PROVEEDOR (CLIENTE) CLIENTE (PROVEEDOR) OQ: COMMISSIONING: PROVEEDOR (CLIENTE) CLIENTE (PROVEEDOR) PQ: ACUERDO DE DISEÑO COMMISSIONING Y CALIFICACIÓN OPERACIÓN CONTINUA (MANTENIMIENTO DEL ESTADO VALIDADO) DE-COMMISSIONING / RETIRO PLAN DE CALIDAD CON CONTRATISTAS ESPECIFICACIÓN DE DISEÑO FUNCIONAL ESPECIFICACIÓN DE PRUEBAS DE ACEPTACIÓN PLAN DE CALIDAD DEL SISTEMA DE PROVEEDORES (5) C&I APLICACIÓN HOJAS DE DATOS
DE DISEÑO C&I APLICACIÓN INGENIERÍA Y DIAGRAMAS ESPECIFICACIÓN DE DISEÑO COMPUTADORA H/W (2) ESPECIFICACIÓN DE PRUEBAS DE
HARDWARE ESPECIFICACIÓN DE DISEÑO S/W
(3) ESPECIFICACIÓN DE PRUEBAS DE
INTEGRACIÓN MODULO DE SOFTWARE PRODUCCIÓN COMPUTADORA H/W ESPECIFICACIÓN DE DISEÑO MODULO S/W (2) ESPECIFICACIÓN DE PRUEBAS MODULO DE SOFTWARE CONFIGURACIÓN / CÓDIGO MODULO S/W
C&I INSPECCIÓN / CALIBRACIÓN (CONTRA PUNTO 5) COMPUTADORA H/W PRUEBAS (CONTRA PUNTO 2) MODULO S/W PRUEBAS (CONTRA PUNTO 4)
MODULO S/W INTEGRACIÓN DE PRUEBAS (CONTRA
PUNTO 4)
SISTEMA DEL PROVEEDOR PRUEBAS DE INTEGRACIÓN (CONTRA PUNTOS 1, 2 Y 3)
DESARROLLO Y DISEÑO
CONSTRUCCIÓN DEL SISTEMA Y DESARROLLO DE
PRUEBAS
REVISIÓN DEL DISEÑO Y PRUEBAS
DE ACEPTACIÓN
PROVEEDOR PROVEEDOR (CLIENTE)
PROVEEDOR PROVEEDOR (CLIENTE)
PROVEEDOR PROVEEDOR (CLIENTE)
PROVEEDOR PROVEEDOR (CLIENTE)
PROVEEDOR PROVEEDOR (CLIENTE)
[image:35.595.78.521.105.683.2]CLIENTE
CAPÍTULO 3. PROPUESTA PARA VALIDACIÓN DE
SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA
INDUSTRIA FARMACÉUTICA.
CAPÍTULO 3.
“PROPUESTA PARA VALIDACIÓN DE
SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO
PARA LA INDUSTRIA FARMACÉUTICA”
Objetivo particular:
Describir mi propuesta de validación de
PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 29 de 125
Capítulo 3. Propuesta para Validación de Sistemas
Automáticos y de Cómputo para la Industria
Farmacéutica.
En el capítulo 2 abordamos los temas de Validación, el Ciclo de Vida como recurso para la validación de sistemas y la GAMP4 como herramienta para la implementación de la metodología para dicha validación. Ahora en este capítulo presento mi propuesta de validación, tomando en cuenta la propuesta de la GAMP4 y el sistema de validación que he llevado en mi experiencia.
3.1 ANÁLISIS.
Desde mi perspectiva la GAMP4 propone 3 caminos para validar los sistemas, ya que marca la diferencia para sistemas IT, para sistemas integrados y para sistemas autónomos.
Mi propuesta consiste en dar un solo esquema de validación general que se pueda aplicar a cualquier clasificación de sistema y que contemple toda la información necesaria para el cumplimiento del Proyecto de Norma Oficial Mexicana y proporcione la base para el cumplimiento regulatorio de otros Países.
La Figura 3.1 muestra mi propuesta para el proceso de validación, donde se pueden observar las fases que propongo en las que se divida la vida de los sistemas y las actividades y/o entregables que se tienen para cada una de ellas.
El alcance de esta propuesta es sólo para los sistemas GMP de Impacto Directo a la calidad del producto.
PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 30 de 125 RESPONSABILIDAD
PROCEDIMIENTOS/ GUIAS/CAPACITACIÓN
ESPECIFICACIÓN DE DISEÑO [a]
PLAN DE VALIDACIÓN ESPECIFICACIÓN DE REQUERIMIENTOS (PRIMERA VERSIÓN) [a] EVALUACIÓN Y SELECCIÓN DEL PROVEEDOR CALIFICACIÓN DE
INSTALACIÓN OPERACIONALCALIFICACIÓN CALIFICACIÓN DE DESEMPEÑO
MANTENIMIENTO DEL SISTEMA DURANTE SU OPERACIÓN RETIRO UBICACIÓN CLIENTE PROVEEDOR PROVEEDOR CLIENTE CLIENTE CLIENTE CLIENTE PROVEEDOR PROVEEDOR CLIENTE PLANEACIÓN Y ESPECIFICACIÓN DISEÑO Y CONSTRUCCIÓN INSTALACIÓN Y PRUEBAS OPERACIÓN DEL SISTEMA RETIRO
PLAN DEL PROYECTO Y CALIDAD
PRUEBAS DE REVISIÓN DE DISEÑO
REPORTE DE VALIDACIÓN Resultados satisfactorios ESPECIFICACIÓN DE REQUERIMIENTOS [b] ESPECIFICACIÓN DE DISEÑO [b]
SI NO MATRIZ DE RASTREABILIDAD Resultados satisfactorios SI NO CORRECCIÓN DEL PROBLEMA ESPECIFICACIÓN DE REQUERIMIENTOS (SEGUNDA VERSIÓN) [a] CLIENTE CLIENTE CLIENTE CLIENTE CLIENTE
PROVEEDOR PROVEEDORCLIENTE
PROVEEDOR PROVEEDOR CLIENTE
CLIENTE
PROVEEDOR PROVEEDORCLIENTE
[image:38.595.82.514.91.660.2]CLIENTE CLIENTE CLIENTE CLIENTE CLIENTE CLIENTE PROVEEDOR CLIENTE PROVEEDOR CLIENTE PROVEEDOR
PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 31 de 125
3.1.1 Correspondencia entre mi Propuesta con el Proyecto de Norma PROY-NOM-059-SSA1-2004, la GAMP4 y NMX-CC-9001-IMNC-2000.
PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
[image:40.595.69.533.136.742.2]Página 32 de 125
Tabla 3.1. Correspondencia entre el Proyecto de Norma PROY-NOM-059-SSA1-2004 y mi Propuesta.
PROY-NOM-059-SSA1-2004 PROPUESTA DE VALIDACIÓN
Equipo automático, mecánico y
electrónico. 10.5
Los sistemas computarizados instalados en los equipos para el control del proceso
de fabricación deben estar validados. 10.5.2 1.2 Marco Normativo.
3.0
Propuesta para Validación de
Sistemas Automáticos y de Cómputo para la Industria Farmacéutica. Con el fin de asegurar la exactitud de los
datos manejados por estos sistemas, se debe implementar un sistema de protección de los mismos para evitar modificaciones a las fórmulas o registros
efectuadas por personal no autorizado. 10.5.3 1.2 Marco Normativo.
Se debe mantener un respaldo en copias fieles, cintas o microfilms, de toda la información archivada en las computadoras o los sistemas relacionados, para asegurar que la información emitida por estos sistemas es exacta, completa y que no existen
modificaciones inadvertidas. 10.5.4 1.2 Marco Normativo.
Validación. 14 1.2 Marco Normativo.
2.1 Validación.
3.0
Propuesta para Validación de
Sistemas Automáticos y de Cómputo para la Industria Farmacéutica.
Política. 14.1 1.2 Marco Normativo.
3.2 Plan de Validación.
Planeación para la validación. 14.2 3.2 Plan de Validación.
Documentación. 14.3 3.2 Plan de Validación.
3.4 Plan del Proyecto y Calidad
3.9 Reporte de Validación
Calificación. 14.4 3.7 Pruebas al Equipo o Sistema.
Sistemas computacionales. 14.8 1.2 Marco Normativo.
Mantenimiento del estado validado. 14.11 3.10 Mantenimiento del Estado Validado
PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
[image:41.595.65.532.124.727.2]Página 33 de 125
Tabla 3.2. Correspondencia entre mi Propuesta y la GAMP4.
PROPUESTA DE VALIDACIÓN GAMP4
Antecedentes y Marco Normativo 1.0 1.0 Introducción a la GAMP (Introduction to GAMP)
Antecedentes 1.1 1.1 ¿Cómo surgió la iniciativa GAMP? (How did the GAMP Initiative start?)
Validación, Ciclo de Vida y GAMP 2.0 6.0 Generalidades de la Validación (Validation Overview)
Validación 2.1
Propuesta para Validación de Sistemas Automáticos y de Cómputo para la
Industria Farmacéutica. 3.0 7.0 Ciclo de Vida de Validación (Validation Life Cycle)
Análisis 3.1 8.0
Sistema de Administración para Proveedores de Sistemas de TI (Management System for Suppliers of IT Systems)
9.0
Validación de Sistemas de Control de Proceso (Process Control System Validation)
Plan de Validación 3.2 7.5 Validación, Plan del Proyecto y Calidad (Validation, Quality and Project Plan)
8.1.2 Planeación (Planning)
9.1 Planeación (Planning)
Especificación de Requerimientos de usuario (URS) y Especificación Funcional
(FS). 3.3 7.3
Especificación de Requirimientos de Usuario (User Requirements Specification)
8.1.3
Especificación, Diseño y Construccción (Specification, Design and
Construction)
9.2 Especificación y Diseño (Specification and Design)
Plan del Proyecto y Calidad 3.4 7.5 Validación, Plan del Proyecto y Calidad (Validation, Quality and Project Plan)
8.1.2 Planeación (Planning)
9.1 Planeación (Planning)
PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 34 de 125 Matriz de Rastreabilidad de
Requerimientos 3.5 8.2.1 Revisiones (Reviews)
9.7 Revisión de Diseño (Design Review)
Especificación de Diseño 3.6 8.1.3
Especificación, Diseño y Construccción (Specification, Design and
Construction)
9.2 Especificación y Diseño (Specification and Design)
Pruebas al Equipo o Sistema 3.7 7.9 Pruebas (Testing)
8.4 Especificaciones de Prueba (Test Specifications)
9.7 Revisión del Diseño (Design Review)
9.12 Desarrollo de Pruebas (Development Testing)
9.13 Pruebas de Aceptación (Aceptance Testing)
9.15 Calificación (Qualification)
Registro de Capacitación 3.8 7.11.2 Capacitación (Training)
8.2.4
Capacitación del Usuario por el
Proveedor (Training of Supplier Staff)
Reporte de Validación 3.9 7.10 Reporte de Validación (Validation Reporting)
9.16 Reporte de Validación (Validation Report)
Mantenimiento del Estado Validado 3.10 7.11 Mantenimiento del Estado Validado (Maintaining the Validated State)
9.17 Mantenimiento del Estado Validado (Maintaining the Validated State)
Retiro 3.11 9.18 Retiro (Retirement)
7.11.14 Retiro del Sistema (System Retirement)
PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
[image:43.595.68.531.137.746.2]Página 35 de 125
Tabla 3.3. Correspondencia entre mi Propuesta y la Norma NMX-CC-9001-IMNC-2000.
PROPUESTA DE VALIDACIÓN NMX-CC-9001-IMNC-2000
Propuesta para Validación de Sistemas Automáticos y de Cómputo para la
Industria Farmacéutica. 3.0
Plan de Validación 3.2 5.4 Planificación
Especificación de Requerimientos de usuario (URS) y Especificación Funcional
(FS). 3.3 5.2 Enfoque al Cliente
7.2.1 Determinación de los requisitos relacionados con el Producto
7.3.2 Elementos de Entrada para el Diseño y Desarrollo
Plan del Proyecto y Calidad 3.4 5.4 Planificación
7.1 Planificación de la realización del Producto
7.3.1 Planificación del Diseño y Desarrollo
7.3.4 Revisión del Diseño y Desarrollo
Matriz de Rastreabilidad de
Requerimientos 3.5 5.2 Enfoque al Cliente
7.2.2 Revisión de los requisitos relacionados con el Producto
7.5.3 Identificación y Trazabilidad
Especificación de Diseño 3.6 7.3 Diseño y Desarrollo
7.3.1 Planificación del Diseño y Desarrollo
7.3.3 Resultados del Diseño y Desarrollo
Pruebas al Equipo o Sistema 3.7 7.2.2 Revisión de los requisitos relacionados con el Producto
7.3 Diseño y Desarrollo
7.3.1 Planificación del Diseño y Desarrollo
7.3.4 Revisión del Diseño y Desarrollo
7.3.5 Verificación del Diseño y Desarrollo
7.3.6 Validación del Diseño y Desarrollo
7.4.3 Verificación de los Productos comprados
Registro de Capacitación 3.8 6.2.2 Competencia, toma de conciencia y formación
Reporte de Validación 3.9 7.3.6 Validación del Diseño y Desarrollo
PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 36 de 125 Mantenimiento del Estado Validado 3.10 4.2.3 Control de los documentos
8.2.2 Auditoría interna
8.2.3 Seguimiento y medición de los procesos
8.2.4 Seguimiento y medición del producto
8.4 Análisis de datos
8.5.1 Mejora continua
8.5.2 Acciones correctivas
PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 37 de 125
3.2 PLAN DE VALIDACIÓN
El primer paso que nos marca la GAMP4 (para los 3 tipos de sistemas), dentro de la etapa de Planeación y Especificación, para iniciar con el ciclo de vida de un sistema es elaborar un Plan de Validación. De hecho nos habla del desarrollo de un Plan Maestro de Validación y de los Planes de validación para proyectos o sistemas.
Un Plan de Validación es el documento donde se definen las actividades de validación, las responsabilidades y los procedimientos a seguir para llevarla a cabo. En cada Empresa dependerá el nombre que se le quiera dar y el alcance que tenga, los más comunes son Plan Maestro de Validación, Plan de Validación y Guías de Validación.
Las siglas en inglés para los términos que utilizaremos para la descripción de estos documentos de validación son: VMP para el Plan Maestro de Validación (Validation Master Plan) y VP para un Plan de Validación (Validation Plan).
Usualmente en el VMP se definen todas las áreas y elementos que requieren validación dentro de una Compañía y se define el plan general para el cumplimiento de esas validaciones específicas. Este Plan no lo comentaremos a fondo ya que el alcance de ese Plan es toda la Planta (limpieza de áreas de producción y equipos, procesos de producción, edificios, instalaciones, equipos y sistemas automáticos y críticos, etc) y nosotros hablaremos del VP que nos definirá como validar en específico uno de los elementos que comprende el VMP.
La importancia del VP radica en que este Plan junto con la Especificación de Requerimientos son la base para definir el Plan del Proyecto y Calidad, entre Usuario y Proveedor.
La GAMP4 nos dice que para cada sistema GMP (Good Manufacturing Practice, Buenas Prácticas de Manufactura) se debe elaborar un Plan de Validación, pero también aclara que cada Compañía establece la estructura en que desarrollara sus Planes.
En esta parte me parece importante mencionar que para un sistema nuevo es conveniente hacer su Plan de Validación individual, pero en el caso de sistemas pequeños que se puedan agrupar o en el caso de aplicar un Plan general de validación para varios equipos iguales en la Planta lo que conviene es hacer un solo Plan que aplique a esos determinados sistemas o equipos.
El Plan de validación es la clave para todo el proceso de validación, en él se puede describir la estrategia de validación que se seguirá o hacer referencia al documento donde está descrito.