Validación concurrente del proceso de fabricación del producto Montelukast comprimidos masticables 5 mg
72
0
0
Texto completo
(2) “No existen límites para forjar tu propia felicidad”.
(3) AGRADECIMIENTOS. En primer lugar, dar las gracias a mi familia por brindarme su apoyo incondicional en esta etapa. He de destacar su esfuerzo, preocupación y sobre todo su interés en cada uno de mis pasos. Por otra parte, entrego toda mi gratitud a Buses Ahumada por financiar mis traslados desde San Felipe, sin ustedes mis posibilidades de estudiar se habrían visto limitadas. Por último, agradezco a Laboratorios Saval por permitirme realizar mi Profundización Profesional y por entregarme una gran gama de conocimientos que me han hecho mejorar como profesional..
(4) LISTA DE ABREVIACIONES AC: Atributo de Calidad ACC: Atributo Crítico de Calidad ANOVA: Analysis Of Variance AV: Valor de aceptación Cp: Índice de Capacidad del Proceso Cpk: Índice de Capacidad Real del Proceso CV: Coeficiente de variación D: Capacidad de Detección DS: Desviación Estándar EPT: Especificación de Producto Terminado F: Frecuencia FMECA: Análisis de Modos de falla, Efectos, y Criticidad (Failur mode, effects and criticality analysis) g: Gramo GMP: Buenas Prácticas de Manufactura traducido al español ISP: Instituto de Salud Pública Kg: Kilogramo L: Litro LEI: Límite Especificación Inferior LES: Límite Especificación Superior M: Masa de la mezcla en gramos Mg: Miligramo mL: Mililitro mm: Milímetro MTK: Montelukast PCC: Parámetro Crítico de Calidad PMW: Plan Maestro de Validación POE: Procedimiento Operativo Estándar PP: Parámetro de Proceso RPN: Risk Priority Number (Número de prioridad de riesgo) S: Severidad SAP: Systems, Applications, Products in data Processing.
(5) SSAM: Área Superficial Específica Media STATA 13: Statistics/Data Analysis 13 VF: Volumen Final ocupado por el sólido en mL.
(6) ÍNDICE GENERAL CONTENIDOS CAPÍTULO I: ESTADO DEL ARTE Y DEFINICIÓN DEL PROBLEMA ...........1 1. Historia de Laboratorios Saval S.A.......................................................... …1 2.Buenas Prácticas de Manufactura (GMP) ....................................................2 2.1 Definición ..........................................................................................2 3.Validación de Procesos................................................................................3 3.1 Requisitos para una Validación de Procesos ...................................3 3.2 Clasificación de Validación de Procesos ..........................................4 3.3 Documentación asociada con la Validación de Procesos .................4 4. Análisis estadístico ......................................................................................6 4.1.Distribución normal ...........................................................................6 4.2.Test Shapiro Wilk .............................................................................7 4.3.Estudio de capacidad .......................................................................8 4.3.1 Índice Cp ...............................................................................8 4.3.2 Índice Cpk .............................................................................9 4.4.Test de Bartlett ...............................................................................10 4.5.Análisis de varianza-ANOVA ..........................................................10 4.6.Cartas de Control ...........................................................................11 4.7.Histograma .....................................................................................12 5.Comprimidos ..............................................................................................13 6.Montelukast................................................................................................14 6.1.Propiedades Físico Químicas .........................................................15 CAPÍTULO II: OBJETIVOS GENERALES Y ESPECÍFICOS .......................16 I.
(7) 1.Objetivo General ........................................................................................16 2.Objetivos Específicos .................................................................................16 CAPÍTULO III: PLAN DE TRABAJO, RESULTADOS Y ANÁLISIS CRÍTICO DE ACUERDO CON CADA OBJETIVO ESPECÍFICO .................................17 1. Actualización del Registro de Fabricación …………………………………..17 1.1 Metodología ....................................................................................17 1.2 Resultados y Discusión ..................................................................18 2.Confección del Protocolo de validación .....................................................19 2.1 Metodología ....................................................................................19 2.1.1 Secciones que componen el Protocolo de Validación .........19 2.1.2 Documentos adjuntos en el Protocolo de Validación ..........20 2.1.2.1 Análisis de Riesgo ...................................................20 2.1.2.2 Diagrama de flujo del Proceso de Fabricación .........21 2.2 Resultados y Discusión ..................................................................22 3. Verificación del Proceso de Fabricación de Montelukast 5 mg Comprimidos Masticables ...................................................................................................25 3.1 Metodología ....................................................................................25 3.1.1 Toma de muestras en las etapas de Pre-lubricación y Lubricación .........................................................................27 3.1.2 Ensayos reológicos………………………………………….….29 3.1.2.1 Densidad……………………………………………...29 3.1.2.2 Granulometría ………………………………………..30 3.1.2.3 Ángulo de reposo…………………………………….31 3.1.3 Toma de muestras en Etapa de Compresión………………..33 3.1.4 Ensayos en Etapa de Compresión……………………….…...34 3.2 Resultados y Discusión…………………………………………………35 II.
(8) 3.2.1 Ensayos en las Etapas de Pre-Lubricación y Lubricación …………………………………………………….35 3.2.2 Ensayos reológicos……………………………………..38 3.2.3 Ensayos en Etapa de Compresión……………………40 4 Confección del Informe de Validación ........................................................43 4.1 Metodología ................................................................................... 44 4.1.1 Secciones del Informe de Validación ..................................44 4.1.2 Análisis estadístico de los datos obtenidos .........................44 4.2 Resultados y Discusión...................................................................45 CAPÍTULO IV: ANÁLISIS CRÍTICO GLOBAL ...............................................51 CAPÍTULO V: CONTEXTUALIZACIÓN DE LOS RESULTADOS EN EL MARCO DE LOS OBJETIVOS DE LA EMPRESA ........................................53 CAPÍTULO VI: CONCLUSIONES Y SUGERENCIAS ...................................54 CAPÍTULO VII: BIBLIOGRAFÍA ....................................................................55. III.
(9) ÍNDICE DE TABLAS Tabla N°1: Valores de Cp y su interpretación…………………………………..9 Tabla N°2: Formulaciones comercializadas de Montelukast, dosis, beneficios y grupos de edad…………………………………………………………………15 Tabla N°3 Estadística descriptiva de los Atributos Críticos históricos y límites de control. ………………………………………………………………………..18 Tabla N°4: Puntuación de RPN establecida por Laboratorios Saval……….21 Tabla N°5: Puntuaciones de RPN para cada etapa del proceso de fabricación de Montelukast 5 mg Comprimidos Masticables……………………………...22 Tabla N°6: Atributos críticos de calidad establecidos en el análisis de riesgo de Montelukast 5 mg comprimidos masticables………………………………23 Tabla N°7: Parámetros Críticos de Calidad (PCC) establecidos en el análisis de riesgo de Montelukast 5 mg comprimidos masticables…………………..24 Tabla N°8: Materias Primas utilizadas en las etapas de Pre-Lubricación y Lubricación……………………………………………………………………….26 Tabla N°9: Equipos e Instrumentos utilizados durante el proceso de Fabricación de Montelukast 5 mg comprimidos masticables………………..26 Tabla N°10: Propiedades de flujo de acuerdo con su ángulo de reposo……32 Tabla N°11: Especificaciones de producto terminado registradas en ISP….34 Tabla N°12: Homogeneidad de granel en Etapa de Pre-Lubricación y Lubricación .…………………………………………………………………….37 Tabla N°13: Ensayos reológicos luego de la Etapa de Lubricación parte 1..39 Tabla N°14: Ensayos reológicos luego de la Etapa de Lubricación parte 2...40 Tabla N°15: Ensayos realizados a los comprimidos de Montelukast 5 mg comprimidos masticables luego de la etapa de Compresión………………..42 Tabla N°16: Estadística descriptiva empleada en los ACC de cada lote productivo. ……………………………………………………………………...45. Tabla N°17: Estadística Descriptiva para el atributo crítico peso luego de la etapa de Compresión……………………………………………………………47 Tabla N°18: Estadística Descriptiva para el atributo crítico dureza luego de IV.
(10) la etapa de Compresión…………………………………………………………47 Tabla N°19: Análisis estadístico Intralote para el ACC peso luego de la etapa de Compresión…………………………………………………………………...48 Tabla N°20: Análisis estadístico Intralote para el ACC dureza luego de la etapa de Compresión……………………………………………………………49 Tabla N°21: Test Shapiro Wilk, ANOVA y Bartlett para los ACC peso y dureza……………………………………………………………………………. 49. V.
(11) ÍNDICE DE FIGURAS Figura N°1: Variabilidad de un proceso por distintos factores involucrados…..6 Figura N°2: Elementos de una carta de control…………………...………...….12 Figura N°3: Estructura de Montelukast sódico (MTK)…………………………15 Figura N°4: Diagrama de Flujo del proceso de Fabricación de Montelukast 5 mg comprimidos masticables…………………………………….………………28 Figura N°5: Puntos de muestreo en mezclador de 300 L……….……………..29. VI.
(12) RESUMEN. La industria farmacéutica para poder comercializar un producto farmacéutico debe asegurar que sus procesos de manufactura estén validados. De esta forma obtienen productos de calidad que cumplen con las normas GMP (Buenas Prácticas de Manufactura, en español) y también con sus clientes. El presente manuscrito tiene como objetivo validar de forma concurrente el proceso de fabricación de un medicamento, que posee como principio activo Montelukast de 5 mg. Para aquello se actualizan y se crean tres registros de fabricación provisorios. Además, se confeccionan protocolo e informe de validación. El tratamiento estadístico adjunto en el Informe de Validación, se basa en la Norma Técnica N°4 de Validación de procesos, descrita por el Instituto de Salud Pública (ISP). A pesar de que las pruebas estadísticas que evalúan la reproducibilidad interlote, Análisis de Varianza (ANOVA) y Test de Bartlett, muestran diferencias estadísticamente significativas para los atributos críticos Peso y Dureza (p-value < 0,05), entre los lotes verificados. Se comprueba empíricamente que el proceso está bajo control y es estable. Los datos están normalmente distribuidos sin la necesidad de efectuar alguna transformación estadística para el atributo peso. En cambio, para el atributo dureza fue necesario realizar un remodelamiento matemático (Boxcox) para lograr la normalidad de los datos y cumplir sistemáticamente con los límites de especificación establecidos para el producto. Los resultados obtenidos para las pruebas realizadas en los distintos lotes productivos: ensayos reológicos, disolución, desintegración, valoración, homogeneidad de granel, peso, dureza, friabilidad y descripción sí cumplen las especificaciones registradas para el producto. Esto indica que es un proceso controlado y reproducible. En definitiva, el proceso de fabricación de Montelukast 5 mg comprimidos masticables se encuentra validado.. VII.
(13) ABSTRACT To market a pharmaceutical product, the pharmaceutical industry must ensure that its manufacturing processes are validated. So, they obtain quality products that comply with GMP standards and also with their customers. The purpose of this manuscript is to concurrently validate the manufacturing process of a medicine that has the Montelukast active ingredient of 5 mg. For that, three provisional manufacturing records are updated and created. In addition, protocol and validation report are prepared. The statistical treatment attached in the Validation Report is based on Technical Standard No. 4 of Validation Process, described by the Public Health Institute (ISP). Even though the statistical tests that evaluate the interlots reproducibility, Analysis of Variance (ANOVA) and Bartlett's Test, show statistically significant differences for the critical attributes weight and hardness (p-value <0.05), among the verified batches. It is empirically verified that the process is under control and is stable. The data is normally distributed without the need to perform any statistical transformation (Boxcox) for the weight attribute. On the other hand, for the hardness attribute, it was necessary to perform a mathematical remodeling to achieve the normality of the data and systematically accomplish with the specification limits established for the product. The results obtained for the tests carried out in the different production batches: rheological tests, dissolution, disintegration, valuation, bulk homogeneity, weight, hardness, friability and description do comply with the specifications recorded for the product. This indicates it is a controlled and reproducible process. In short, the manufacturing process of 5 mg Montelukast chewable tablets is validated.. VIII.
(14) CAPÍTULO I: ESTADO DEL ARTE Y DEFINICIÓN DEL PROBLEMA. 1. Historia de Laboratorios Saval S.A Laboratorios Saval inicia su trayectoria en 1939 en España, ligado de manera exclusiva al campo de la oftalmología. Luego, en la década de los 40 comienza la producción de medicamentos en Chile, principalmente en la fabricación de productos oftálmicos. Al paso de los años se amplía su participación en otras especialidades y el desarrollo de nuevas formas farmacéuticas, ofreciendo una alta gama de productos y servicios que contribuyen al bienestar de las personas. Esto ha permitido situar a la empresa en un nivel de liderazgo dentro de la industria farmacéutica nacional y la comercialización a distintos países, mayoritariamente en Latinoamérica [3]. Por otra parte, Laboratorios Saval en el año 2000 fue el primer laboratorio en Chile que recibió certificación del cumplimiento de las normas GMP por parte del ISP (Instituto de Salud Pública). Cabe destacar que lo hizo cinco años antes de que fuera un requisito obligatorio para la industria farmacéutica chilena [3]. Laboratorios Saval posee modernas instalaciones, donde se desarrollan y producen distintas formas farmacéuticas, organizados en dos edificios. El primero, está destinado para la producción exclusiva de fármacos penicilínicos sólidos: comprimidos, comprimidos recubiertos, comprimidos dispersables y polvo para suspensión. Mientras que, el segundo edificio está designado para la producción de distintos productos. [4]. :. - Productos Líquidos: jarabes, suspensiones y gotas orales. - Productos Semisólidos: cremas, geles dérmicos y supositorios. - Productos Estériles: inyectables y productos oftálmicos.. 1.
(15) - Productos. Sólidos:. comprimidos,. comprimidos. recubiertos,. comprimidos dispersables, cápsulas, polvo para suspensión y comprimidos recubiertos. El proyecto de profundización profesional se lleva a cabo en el área de sólidos, donde se realiza una validación concurrente del proceso de fabricación del producto Montelukast 5 mg comprimidos masticables. Para aquello, se coordina con el área de producción para el seguimiento a cada uno de los procesos involucrados.. 2. Buenas Prácticas de Manufactura (GMP) 2.1 Definición Actualmente, todos los medicamentos de uso humano fabricados e importados en Chile, e incluso los exportados, deben cumplir con las directrices y principios de las GMP. Las GMP son normas técnicas mínimas establecidas para todos los procesos involucrados en la fabricación de productos farmacéuticos, destinadas a garantizar la calidad de estos. Determinada por su eficacia, seguridad y estabilidad, conforme a las características de identidad, potencia, pureza, entre otras características establecidas en sus respectivos registros sanitarios [1]. Uno de los principios que establece las GMP expone que las industrias farmacéuticas. para. asegurar. la. entrega. y. comercialización. de. medicamentos con altos estándares de calidad, deben validar todos sus procesos de producción. [3]. . De esta forma se asegura que los productos. farmacéuticos poseen procesos consistentes y controlados de acuerdo con los criterios de calidad apropiados para su uso. Además, se señala que el cumplimiento de estas permite obtener un producto capaz de cumplir con todas las especificaciones de calidad 2 1.
(16) previstas en su diseño de manera reproducible y, entregar validez a los estudios de bioequivalencia. [5,6]. .. 3. Validación de Procesos Una validación de procesos ha sido definida según la Guía Técnica G-Biof 02 descrita por el ISP como la “obtención de la evidencia documentada, que provee. un. alto. nivel. de. seguridad,. que. un. proceso. específico. consistentemente resultará en un producto que cumple sus especificaciones predeterminadas y características de calidad” [6]. El objetivo de una Validación de procesos es demostrar y proporcionar de forma continua y reproducible productos farmacéuticos que cumplan con sus especificaciones de calidad y diseño. Para lograr este objetivo los laboratorios deben planificar la validación de manera que garanticen el cumplimiento regulatorio y asegurando que la calidad, seguridad y consistencia del producto no se encuentren comprometidas. [1]. .. 3.1 Requisitos para una Validación de Procesos Una validación de procesos debe cumplir con algunos requisitos previos, los cuales deben estar debidamente documentados según lo exigen las GMP. Dentro de los requisitos exigidos se detalla - Soporte. documental. adecuado,. [7]. :. sistema. de. documentación. controlado. Procedimientos, formatos, protocolos, etc. - Proveedores calificados. - Áreas y equipos calificados y calibrados (vigentes). - Validación de limpieza (áreas y equipos). - Validación de metodologías analíticas. - Plan maestro de validación definido y autorizado. - Estandarización de la producción. 3.
(17) - Personal capacitado y calificado. 3.2 Clasificación de Validación de Procesos Actualmente existen distintos tipos de Validaciones de Proceso. En primer lugar, está la Validación Prospectiva, la cual es completada antes de la fabricación del producto final, se lleva a cabo durante la etapa de desarrollo controlado con base a un análisis de riesgo del proceso de producción, el que se desglosa en pasos individuales; estos luego son evaluados. en. base a la experiencia pasada para determinar si pueden llevar a situaciones críticas [5]. En segundo lugar, es posible validar proceso de fabricación durante su proceso productivo, a este se le denomina Validación Concurrente, es decir, se realiza durante la producción de rutina de productos destinados a ser comercializados [5]. En tercer lugar, se encuentra la Validación Retrospectiva donde los procesos que han sido utilizados por bastante tiempo y sus procedimientos, equipos y composición se han mantenidos inalterados, pueden ser validados con la revisión y análisis de los datos documentados de fabricaciones anteriores [5]. Por último, los productos farmacéuticos pueden ser validados por medio de una Revalidación para asegurar que no existe alguna variación en el proceso que pueda afectar las características de calidad del producto resultante y dar cumplimiento con los requisitos establecidos [5]. 3.3 Documentación asociada con la Validación de Procesos La información recolectada, analizada y revisada durante una Validación de Procesos se basa en los criterios de aceptación definidos previamente por cada Industria Farmacéutica. Sin embargo, la entidad regulatoria les exige presentar una serie de documentos para sus respectivas Validaciones de Procesos [5]. Entre ellos destacan: 4.
(18) - Procedimientos operativos estándar (POE): Corresponden a un documento escrito y autorizado que contiene todas las instrucciones específicas, por ejemplo, operación de equipo, muestreo, limpieza de instalaciones, uso y manejo de equipos, entre otros. Algunos de estos pueden ser usados para complementar la documentación requerida en la validación de un lote productivo. [5]. .. - Plan maestro de validación (PMW): Documento de alto nivel que establece los elementos claves del programa de validación. Resume de forma concisa y clara toda la información del trabajo de validación del. laboratorio;. instalaciones,. sistemas,. equipos,. formato. de. documentación, planificación y calendarización, control de cambios e incluso una declaración de responsabilidad de todos los participantes que implementan el plan. [5]. .. - Protocolo de validación: Documento que incluye todas las actividades a desarrollar durante la validación de procesos, junto con los criterios de aceptación para considerar un proceso de fabricación exitosa. Como mínimo debe incluir, objetivos, POE a seguir, equipos utilizados, estándares de aceptación, el tipo de validación, descripción de los procesos y/o parámetros, requisitos de muestreo, etc. [5]. - Informe de Validación: Documento que incluye registros, resultados y la evaluación de la validación. Además, puede incluir optimización del proceso y/o equipos. Se requiere que incluya un título, objetivos, detalle de las materias primas y equipos utilizados, procedimientos y métodos de prueba. Por otra parte, los resultados deben ser evaluados, analizados y comparados con criterios de aceptación. Si existiese una desviación durante la validación de procesos debe ser abordada y justificada. Por último, debe incluir una conclusión donde se dé a conocer si la validación de procesos fue exitosa o no. [5]. .. 5 4.
(19) 4. Análisis estadístico Es fundamental en una validación de procesos un análisis estadístico de los datos obtenidos en las diferentes mediciones realizadas durante el proceso de fabricación para evaluar los atributos críticos de proceso. [2]. .. Un proceso productivo está sometido a una serie de factores que hacen imposible fabricar dos productos exactamente iguales (Figura 1). Se ha visto que las características del producto presentan una cierta variabilidad (mano de obra, equipos, materiales, método, ambiente, mantenimiento, etc.). La cual es indeseable y el objetivo a seguir es disminuirla o al menos mantenerlas dentro de ciertos límites. [8]. .. Para alcanzar este objetivo el control estadístico es una herramienta que contribuye a una mejora en la calidad de la fabricación y permite obtener un mayor conocimiento acerca del proceso.. Figura 1: Variabilidad de un proceso por distintos factores involucrados [8]. 4.1. Distribución normal La distribución normal es probablemente la distribución continua más importante, tanto en estadística teórica como aplicada. Al graficar en función 6.
(20) de su densidad se obtiene una forma de campana (unimodal), simétrica a ambos lados de la media. [8]. .. En una serie de valores que sigan una distribución normal, se encontrará que; entre ±1 desviaciones estándar de la media se ubican el 68,27% de los datos, entre ±2 desviación estándar de la media se ubican el 95,45% y entre ±3 desviaciones estándar de la media se ubican el 99,73% de la distribución [8]. .. Muchos análisis estadísticos son susceptibles de producir resultados erróneos si se aplican a un conjunto de datos que no siga una distribución normal [9]. Por esta razón, es indispensable verificar que los datos obtenidos se correlacionen con dicha distribución, antes de proseguir con el tratamiento de datos. Sin embargo, mediante el programa estadístico detallado más abajo es posible hacer un remodelamiento matemático (Boxcox), el cual entrega un coeficiente de elevación óptimo para lograr la conversión de los datos. 4.2. Test Shapiro Wilk Test de Shapiro Wilk ejecutado en el programa STATA 13 (Statistics/Data Analysis) permite comprobar la normalidad de un conjunto de datos. [9]. .. Este test, acorde al software utilizado, plantea las siguientes hipótesis: - Hipótesis Nula (H0): Los datos seleccionados pertenecen a una población que sigue una distribución estadística normal. - Hipótesis alternativa (H1): Los datos seleccionados NO pertenecen a una población que siga una distribución estadística normal. Dónde: - Si el p-value es mayor a 0,05 se acepta H0 - Si el p-value es menor a 0,05 se rechaza H0. 7.
(21) 4.3. Estudio de capacidad La capacidad de un proceso corresponde a un atributo medible que puede obtenerse por medio del índice de capacidad el proceso (Cp, Cpk), esta puede analizarse de forma estadística, siempre que los datos obtenidos muestren una distribución normal. [2]. .. Los índices de capacidad son estimaciones numéricas de la capacidad de proceso, es decir, nos dan una idea de qué tan capaz es el proceso y a qué nivel podría cumplir con las especificaciones. Estos son muy útiles y fáciles de calcular, no tienen unidades de medida, por lo que permiten comparar distintos procesos [8]. 4.3.1 Índice Cp Básicamente, el Índice de Capacidad del Proceso (Cp) establece una relación entre los límites de especificación (variación tolerada) y la variación natural o real del proceso. 𝐶𝑝 = (𝐿𝐸𝑆 − 𝐿𝐸𝐼)/6𝐷𝑆. Dónde: LES= Límite especificación superior LEI= Límite especificación inferior DS= Desviación estándar En la fórmula de Cp se incluye seis veces la desviación estándar que corresponde a la variación real del proceso. En donde se afirma que entre media ± 3DS se encuentra el 99.73% de los valores de una variable con distribución normal. [8]. .. Para que un proceso sea considerado potencialmente capaz de cumplir con las especificaciones se requiere que la variación real sea menor que la variación tolerada. Además, es deseable que el valor de Cp sea mayor a 1, por el contrario, no se cumpliría con estas. Para una mayor interpretación 8.
(22) en la Tabla N°1 se presenta una clasificación de Cp de acuerdo con el valor calculado [8]. Tabla N°1: Valores de Cp y su interpretación. 1<. [8]. .. Valor de Cp. Clase del proceso. Decisión. Cp ≥ 2. Clase mundial. Cp >1.33. 1. Adecuado. Cp < 1.33. 2. Parcialmente adecuado, requiere de un control estricto. 0.67< Cp < 1. 3. No adecuado, es necesario un análisis del proceso. Requiere de modificaciones serias para alcanzar la calidad satisfactoria. Cp < 0.67. 4. No adecuado. Requiere modificaciones muy serias.. Se tiene calidad de Seis Sigmas. de. 4.3.2 Índice Cpk El índice Cpk se conoce como el índice de capacidad real del proceso, es considerado la versión mejorada de Cp, que sí toma en cuenta el centrado del proceso. Una de las formas más comunes de calcularlos es la siguiente: 𝑚𝑒𝑑𝑖𝑎 − 𝐿𝐸𝑆 𝐿𝐸𝑆 − 𝑚𝑒𝑑𝑖𝑎 𝐶𝑝𝑘 = 𝑚í𝑛𝑖𝑚𝑜[ , ] 3𝐷𝑆 3 𝐷𝑆. El índice Cpk siempre será menor o igual al valor de Cp. Cuando están muy próximos indica que la media está muy cerca del punto medio de las especificaciones, por lo que la capacidad potencial y real son similares. [8]. .. Cuando el valor de Cpk sea mayor a 1.25 en un proceso ya existente, se considerará que el procedimiento tiene un proceso con capacidad satisfactoria. Mientras que en un proceso nuevo el valor de Cpk debe ser mayor a 1.45. Por el contrario, si se obtiene un Cpk < 1 el proceso no cumple con por lo menos una especificación [8].. 9.
(23) 4.4. Test de Bartlett El test de Bartlett permite comprobar si las muestras de las poblaciones en estudio poseen la misma varianza. Aquellas que poseen varianzas iguales se les denominan homocedasticidad de varianzas entre los distintos lotes en estudio [10]. Como es una de las suposiciones que requiere el análisis de varianza, este test debe realizarse antes de la prueba ANOVA. Las hipótesis asociadas a este test son las siguientes: -. Hipótesis Nula (H0): Las varianzas poblacionales de los datos. seleccionados no presentan diferencias estadísticas significativas. -. Hipótesis Alternativa (H1): Las Varianzas poblacionales de los. datos seleccionados presentan diferencias estadísticas significativas. Dónde: -. Si el p-value es mayor a 0,05 se acepta H0. -. Si el p-value es menor a 0,05 se rechaza H0. 4.5. Análisis de varianza-ANOVA El análisis de varianza es un método para probar la igualdad de dos o más medias poblacionales. Este método utiliza la distribución de probabilidad continua F, o también conocida como F de Snedecor. El análisis de ANOVA se basa en la comparación de dos estimaciones: la varianza intralote e interlote. Para aquello es necesario aceptar las siguientes suposiciones [10]: -. Las poblaciones de las muestras recolectadas poseen una. distribución normal. -. Las poblaciones de las muestras recolectadas poseen la misma. varianza, previamente verificada con el test de Barttlet. 10.
(24) -. Las. muestras. fueron. recolectadas de. forma. aleatoria. e. independiente desde sus respectivas poblaciones. Este test se puede utilizar cuando se quiere evaluar una característica o factor, denominado “análisis de varianza de una vía” que considera las siguientes hipótesis [10]: -. Hipótesis Nula (H0): Las medias poblacionales de los datos. seleccionados no presentan diferencias estadísticas significativas. -. Hipótesis alternativa (H1): Las medias poblacionales de los datos. seleccionados presentan diferencias estadísticas significativas. Dónde: -. Si el p-value es mayor a 0,05 se acepta H0. -. Si el p-value es menor a 0,05 se rechaza H0. También, es posible usar un test equivalente, con el enfoque de la región crítica, que utiliza el estadístico F. De esta manera, mediante el módulo de análisis estadístico del software Microsoft Excel, se obtiene un valor F calculado y un valor F crítico, los cuales se interpretan así -. [10]. :. Si F (calculado) es mayor a F (crítico) significa que SÍ hay una. diferencia estadística significativa entre los grupos (filas o columnas). -. Si F (calculado) es menor a F (crítico) significa que NO hay una. diferencia estadística significativa entre los grupos (filas o columnas). 4.6. Cartas de Control El objetivo de una carta de control es observar y analizar el comportamiento de un proceso productivo a través del tiempo. Así, es posible distinguir las variaciones por causas comunes y/o atribuibles. Esto permite caracterizar el funcionamiento del proceso y decidir las mejores acciones de control y mejora [8]. Las cartas de control contienen límites de control que definen el inicio y final 11.
(25) del rango de variación. Debe quedar claro que estos no son las especificaciones, corresponden a un intervalo mucho más acotado (figura 2) [8]. En un proceso de fabricación óptimo todos los datos obtenidos deben caer dentro de esos límites y estar cercanos a la línea central. Si se observa un punto fuera de los límites de control, es señal de que ocurrió algo fuera de lo usual en el proceso [8].. Figura 2: Elementos de una carta de control [8]. 4.7. Histograma El histograma es una representación gráfica en forma de barras, resultado de un número suficiente de datos (de preferencia mayor a 100) de la distribución de un conjunto de datos o una variable, donde los datos son agrupados según su magnitud. En la gráfica cada barra representa un grupo, en que su altura es proporcional a la frecuencia de los valores representados. Generalmente, el eje vertical corresponde a la frecuencia de los datos mientras que el eje horizontal representa la magnitud de estos [8]. Por medio de un histograma es posible ver la tendencia central de los datos, 12.
(26) facilitar el entendimiento de la variabilidad y favorecer el pensamiento estadístico. Puesto que, nos podría indicar una idea acerca de la capacidad del proceso [8]. 5. Comprimidos Actualmente, los comprimidos constituyen la forma farmacéutica más utilizada, representan entre el 40% y el 70% de todas las formas de dosificación. Generalmente, están diseñados para ser deglutidos y luego ser absorbidos a nivel gastrointestinal. [11]. .. Los comprimidos pueden variar en lo relativo a su forma, tamaño y peso. El tamaño suele oscilar entre 5 y 17 mm (milímetros) mientras que su peso entre 0,1 y 1,0 g (gramos), dependiendo de la dosis del principio activo, sus características y el uso de estos [11]. Actualmente, existen diversos tipos de comprimidos que se clasifican de la siguiente manera según la Guía para la denominación de los productos farmacéuticos en Chile [12]: - Comprimidos no recubiertos - Comprimidos recubiertos - Comprimidos efervescentes - Comprimidos solubles - Comprimidos dispersables/masticables/sublinguales - Comprimidos bucodipersables - Comprimidos. recubrimiento. de. liberación. entérico. modificada:. (Comprimidos. comprimidos. gastroresistentes. con o. de. liberación retardada) En el caso de los comprimidos masticables están destinados a ser fragmentados en la boca y posteriormente deglutidos. Se caracterizan por 13 12.
(27) no poseer disgregantes ni recubrimiento dentro de su formulación, incluir ciertos saborizantes y aromatizantes. Están pensados principalmente para pacientes que presenten dificultades de deglución y para niños. [11,12]. .. Por otra parte, un producto farmacéutico terminado debe cumplir con ciertas especificaciones. Estas son parte fundamental de un Registro Sanitario, que constituye un elemento de garantía de calidad de los medicamentos. [13]. .. Las formas farmacéuticas sólidas deben cumplir con los siguientes ensayos: - Disolución (aparato/velocidad) - Friabilidad (Límite máximo, %) - Uniformidad de dosis unitaria (% de lo declarado) - Control de peso (Fórmula (mg) y límites) - Descripción (Aspecto, color, forma, etc.) - Dimensiones (Valor teórico en mm de diámetro espesor, largo, ancho) - Dureza (Valor teórico y límites) - Valoración del (los) principio (s) activo (s) (Valor teórico, método) - Ensayos de sustancias relacionadas, impurezas o productos de. degradación si procede - Tipo y material de envase. 6. Montelukast Montelukast es un principio activo de creciente interés como terapia alternativa para el asma en diferentes grupos de edad (Tabla N°2) debido a sus propiedades bronco-protectoras, antiinflamatorias y antialérgicas. Actualmente, es administrado sólo por vía oral, que puede plantear un problema en la administración en niños de corta edad o pacientes con disfagia [14].. 14.
(28) Tabla N°2: Formulaciones comercializadas de Montelukast, dosis, beneficios y grupos de edad. [14]. .. Dosificación. Formulación. Beneficios. Grupo de edad. 4 mg. Gránulos orales. Fácil de tragar, dispersable y compatible con la leche materna. 6 meses a 5 años. 4 mg. Comprimidos masticables. Adecuado para jóvenes. 2 a 5 años. 5 mg. Comprimidos masticables. Adecuado para jóvenes. 6 a14 años. 10 mg. Comprimidos recubiertos. Resistente a la luz de día. ≥15 años. 6.1. Propiedades Físico Químicas Montelukast (Figura 3) posee ciertas limitaciones para su uso y formulación asociadas a sus propiedades fisicoquímicas, ya que tanto en su forma neutra como de sal, es sensible a la luz, temperatura, humedad y oxidación. En este contexto, las formas farmacéuticas disponibles en el mercado están diseñadas para disminuir la exposición a la luz y a la humedad, lo cual explica por qué las formulaciones líquidas aún no son comercializadas. [14]. .. Figura 3: Estructura de Montelukast sódico (MTK) [15]. 15.
(29) CAPÍTULO II: OBJETIVOS GENERALES Y ESPECÍFICOS. 1. Objetivo General Validar de forma concurrente el proceso de fabricación del producto Montelukast comprimidos masticables 5 mg, elaborados en el área de fabricación de sólidos de Laboratorios Saval.. 2. Objetivos Específicos. I.. Actualizar Registro de Fabricación del producto de acuerdo con el proceso de fabricación estandarizado.. II.. Confeccionar Protocolo de validación del producto acorde a las características a evaluar en la Validación de Procesos y que sea afín con el registro de fabricación.. III.. Verificar el proceso de fabricación de tres lotes productivos siguiendo el Registro de Fabricación.. IV.. Confeccionar Informe de Validación del producto acorde a los resultados obtenidos y la evaluación estadística, según la exigencia del ISP.. 16.
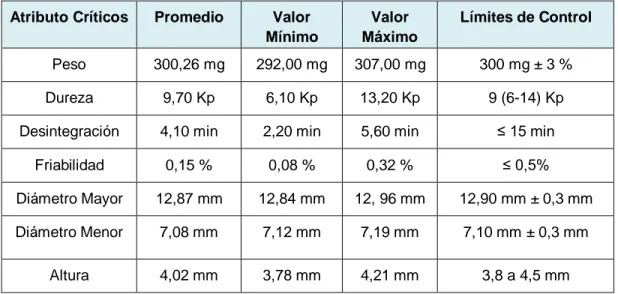
(30) CAPÍTULO III: PLAN DE TRABAJO, RESULTADOS Y ANÁLISIS CRÍTICO DE ACUERDO CON CADA OBJETIVO ESPECÍFICO La validación del proceso de fabricación del producto Montelukast 5 mg comprimidos masticables es de tipo concurrente, para aquello se fabricaron tres lotes industriales a una escala de 220.000 comprimidos, equivalente a 66 Kg. 1. Actualización del Registro de fabricación 1.1 Metodología En primera instancia, el registro de fabricación vigente para el producto a validar se bloqueó del sistema informático SAP (System, Applications, Products in data Processing) mediante solicitud al Departamento de Aseguramiento de calidad. Luego, se realizó una revisión histórica de 50 lotes productivos. En la cual se evaluaron los resultados históricos de los atributos de Calidad (AC); peso, dureza, dimensiones del comprimido, desintegración y friabilidad. Posteriormente, se realizó un análisis estadístico de tipo descriptivo para cada atributo y se comparó con lo descrito en el registro de fabricación con el fin de mantener o editar los límites de control en la etapa de compresión. En cambio para las etapas de pre-lubricación y de lubricación se evaluó los datos de homogeneidad de Montelukast en los respectivos graneles, en la misma cantidad de lotes fabricados previamente. A continuación, se analizaron los resultados para ver si eran conformes de acuerdo con la especificación interna del laboratorio (85%-115%), para ajustar y/o mantener los parámetros de proceso (PP) en los pasos de mezclado y tamizado Para la etapa de compresión se decidió caracterizar los PP durante la fabricación de los lotes productivos, por ello no se detallaron datos al interior del registro de manufactura.. 17.
(31) 1.2 Resultados y Discusión Al realizar una revisión histórica de los AC de 50 lotes productivos para la etapa de compresión, se decidió mantener los límites de control descritos en el registro de fabricación anterior. De acuerdo con la Tabla N°3, tanto la media como los valores mínimos y máximos están dentro de estos rangos. Además, al establecer límites de control más estrechos que los de especificación, se aseguró el cumplimiento de estos últimos que suelen ser más amplios. Si llegase a ser necesario modificar algunos de estos AC se evaluará en el transcurso de la fabricación de los tres lotes destinados a la validación de procesos. Tabla N°3: Estadística descriptiva de los Atributos Críticos históricos y límites de control. Atributo Críticos. Promedio. Valor Mínimo. Valor Máximo. Límites de Control. Peso. 300,26 mg. 292,00 mg. 307,00 mg. 300 mg ± 3 %. Dureza. 9,70 Kp. 6,10 Kp. 13,20 Kp. 9 (6-14) Kp. Desintegración. 4,10 min. 2,20 min. 5,60 min. ≤ 15 min. Friabilidad. 0,15 %. 0,08 %. 0,32 %. ≤ 0,5%. Diámetro Mayor. 12,87 mm. 12,84 mm. 12, 96 mm. 12,90 mm ± 0,3 mm. Diámetro Menor. 7,08 mm. 7,12 mm. 7,19 mm. 7,10 mm ± 0,3 mm. Altura. 4,02 mm. 3,78 mm. 4,21 mm. 3,8 a 4,5 mm. Luego, para las etapas de pre-lubricación y lubricación se decidió dejar los mismos parámetros de proceso para los pasos de tamizado y mezclado, dado que los valores obtenidos anteriormente de la homogeneidad de los respectivos graneles presentan un promedio de 94,5%. Esto indicó que los valores previos son conformes de acuerdo con la especificación definida por el laboratorio (85%-115%). 18.
(32) Los parámetros de proceso tales como tiempos de mezclado, apertura de malla y orden de adición de los componentes no se detallaron por confidencialidad con el laboratorio. Además, se decidió incluir en los registros provisorios de fabricación avisos para toma de muestras para las etapas de pre-lubricación, lubricación y compresión. También, se colocaron tablas para anotar los parámetros de proceso empleados en la manufactura del producto. Aquello facilitó la toma de datos durante el proceso y el correcto seguimiento de los pasos por parte del operador. De acuerdo con lo mencionado anteriormente se modificó el método de fabricación y se confeccionaron tres registros provisorios que fueron utilizados durante la verificación del proceso. Cabe mencionar que una vez finalizada la validación de proceso se creó una nueva versión para el método de manufactura, que incluyó todos los parámetros asociados a las distintas etapas del proceso de fabricación para controlar los lotes productivos post-validación. 2. Confección del Protocolo de validación 2.1 Metodología 2.1.1 Secciones que componen el Protocolo de Validación El Protocolo de Validación se dividió de la siguiente forma de acuerdo con el POE del laboratorio: -. Introducción. -. Objetivos. -. Alcance. -. Responsabilidades. -. Descripción general del proceso (Fórmula y Proceso de Fabricación). -. Metodología de validación 19.
(33) -. Desviaciones. -. Documentos relacionados y referencias. -. Control de cambios. -. Anexos (Pruebas y ensayos para la validación, Diagrama de Flujo y. Análisis de Riesgo). 2.1.2 Documentos adjuntos en el Protocolo de Validación 2.1.2.1 Análisis de Riesgo Se diseñó un análisis de riesgo para cada etapa del proceso de fabricación de Montelukast 5 mg comprimidos masticables, en el cual los atributos críticos de calidad se relacionaron con la seguridad, efectividad, pureza y potencia del producto. En primer lugar, se hizo un análisis FMECA (Failur Mode, Effects and Criticality Analysis) para identificar los parámetros críticos involucrados que deben ser controlados durante el proceso, para asegurar la reproducibilidad de los atributos críticos de calidad. Luego, para evaluar los posibles riesgos que puedan generar un incumplimiento de cada atributo de calidad, se evaluó su criticidad por medio del impacto que tendría sobre las relaciones y seguridad del consumidor, efecto sobre el uso, pérdidas para la empresa y efecto sobre las regulaciones gubernamentales. Los atributos de calidad (AC) que sean considerados críticos se nombran atributos críticos de calidad. Posteriormente, se utilizó un diagrama de causa y efecto conocido como la espina de pescado. Este diagrama se centró en cinco grupos que pueden ser las posibles causas: hombre, máquina, entorno, material y método o medida. A cada una de estas causas se evaluó su severidad (S), frecuencia de ocurrencia (F) y Capacidad de detección (D). Para aquello, se les asignó una puntuación de 1 a 10 a cada una, el valor menor representa una menor probabilidad de causalidad mientras que un valor más cercano a 10 indicaría una mayor probabilidad de riesgo en el proceso de fabricación. 20.
(34) Por consiguiente, se realizó un puntaje final que corresponde a la multiplicación entre S, F y D, denominada Número de prioridad de riesgo (RPN). Para evaluar si una causa es crítica se utilizó la clasificación de la Tabla N°4 establecida de forma arbitraria en Laboratorios Saval. Comúnmente una causa con una puntuación mayor a 96 se considera crítica. Tabla N°4: Puntuación de RPN establecida por Laboratorios Saval Puntaje de resultados 1 - 49. Riesgo bajo, no requiere acción. 50 - 99. Riesgo medio, requiere acción. ≥ 100. Riesgo Alto, requiere acción y verificación del riesgo. 2.1.2.2 Diagrama de flujo del Proceso de Fabricación Posteriormente, se elaboró un diagrama de flujo de todo el proceso de fabricación para tener una visión global de este, junto con las variables de entrada. (parámetros. de. proceso,. materias. primas,. documentación. involucrada, etc.) y las de salida que corresponden a los atributos de calidad medibles. Además, aquí se detalló la metodología, equipos e instrumentos utilizados y el producto resultante de cada etapa. La hoja correspondiente a cada etapa de fabricación se dividió en tres columnas. La primera ubicada hacia la izquierda, incluyó la documentación, materiales de partida y parámetros del proceso a controlar. Por otro lado, la columna del centro contenía todos los pasos de fabricación de forma ordenada, breve y concisa. Finalmente, en la columna derecha se colocó todos los atributos de calidad medibles, con metodologías analíticas e instrumentos a utilizar en cada uno.. 21.
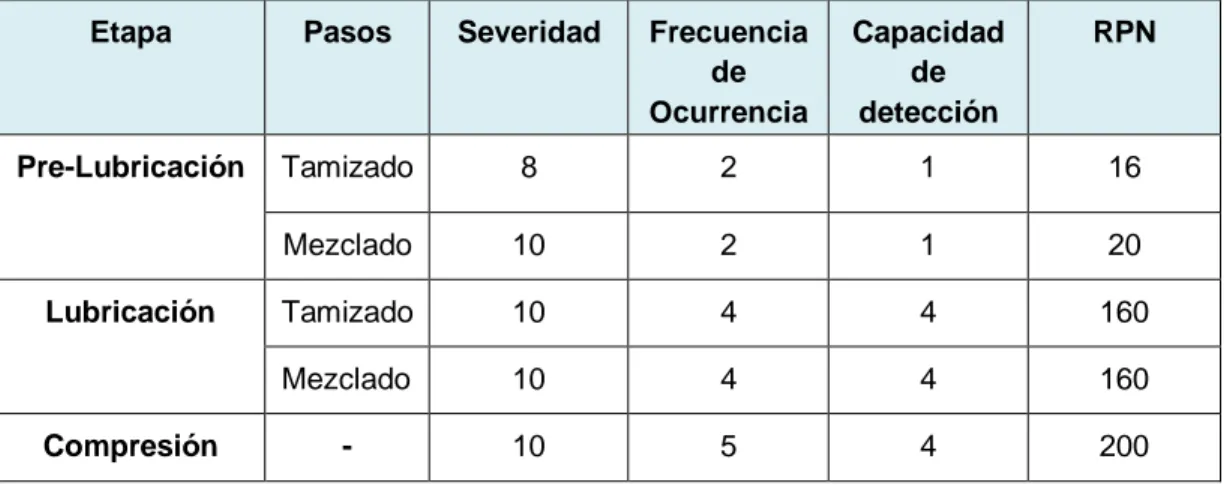
(35) 2.2 Resultados y Discusión Dentro del análisis de riesgo realizado para el proceso de fabricación de Montelukast 5 mg comprimidos masticables, se determinó que existen dos etapas críticas dentro del proceso de manufactura, lubricación y compresión, según la puntuación obtenida en el análisis causa y efecto. La Tabla N°5 da cuenta de los valores de Severidad (S), Frecuencia de ocurrencia (O) y Capacidad de detección (D) asignados para cada una de las etapas de acuerdo con la experiencia de la fabricación del producto y de forma arbitraria. A partir de estas se desprendió un RPN mayor a 100 en las etapas de Lubricación y Compresión, lo cual reflejó que son etapas de alto riesgo, que requieren un seguimiento del proceso y verificación de este. Dado que la puntuación RPN es muy subjetiva, se sugiere implementar una matriz de criticidad para cada una de las causas (S, F y D) involucradas. Así se lograría una estandarización a la hora de estimar la puntuación. Pese a no considerar la etapa de pre-lubricación como crítica, se debe tener precaución en ocupar la malla correcta y respetar el orden de adición de las materias primas en los pasos de tamizado. De la misma forma en los pasos de mezclado se sugiere respetar los tiempos de mezclado para lograr la correcta homogeneización del principio activo. Tabla N°5: Puntuaciones de RPN para cada etapa del proceso de fabricación de Montelukast 5 mg Comprimidos Masticables. Etapa. Pasos. Severidad. Frecuencia de Ocurrencia. Capacidad de detección. RPN. Pre-Lubricación. Tamizado. 8. 2. 1. 16. Mezclado. 10. 2. 1. 20. Tamizado. 10. 4. 4. 160. Mezclado. 10. 4. 4. 160. -. 10. 5. 4. 200. Lubricación. Compresión. 22.
(36) Por otra parte, el análisis de riesgo confeccionado permitió identificar los Atributos Críticos de Calidad para cada etapa crítica, detallados en la Tabla N°6. Se consideraron como Atributos Críticos de Calidad todos los test que se realizan en las etapas de Lubricación y Compresión, que están registrados en las especificaciones de producto terminado. Además, se analizó el impacto que tendría sobre las relaciones con el consumidor, su uso, pérdidas para la empresa, etc. Tabla N°6: Atributos críticos de calidad establecidos en el análisis de riesgo de Montelukast 5 mg comprimidos masticables. Etapa de Proceso. ACC. Lubricación. Densidad aparente y consolidada, granulometría, ángulo de reposo y homogeneidad de granel.. Compresión. Peso, dureza, desintegración, friabilidad, valoración, disolución y uniformidad de dosis.. Dado que los ACC pueden verse afectados por los parámetros de proceso, se realizó un análisis FMECA y se identificaron las posibles causas que se deben vigilar con mayor prioridad para mantener controlado los ACC descritos en la Tabla N°7.. Para la etapa de lubricación se debió verificar el. cumplimiento de parámetros de los pasos de tamizado y mezclado, mientras que para la etapa de compresión se verificaron los parámetros propios de la tabletera asociado con el peso, dureza y dimensiones de cada comprimido.. 23.
(37) Tabla N°7: Parámetros Críticos de Calidad (PCC) establecidos en el análisis de riesgo de Montelukast 5 mg comprimidos masticables. Etapa de Proceso. PCC. Lubricación. Apertura de malla, orden de adición de los componentes, tiempo de mezclado y velocidad de mezclado. Compresión. Velocidad de compresión, ajuste de peso y dureza mediante parámetros de tabletera.. Luego de evaluar las etapas críticas del proceso se realizó la confección del protocolo de validación, el cual no sufrió grandes modificaciones al comparar con el formato propio del laboratorio. En este se completó toda la información requerida respecto a las pruebas y ensayos que se emplearon para comprobar la reproducibilidad de los lotes que fueron validados. Este incluyó todas las operaciones a realizar, el análisis estadístico involucrado, atributos críticos de calidad, parámetros de procesos y método de manufactura vigente. Además, contenía un apartado de pre-requisitos que incluyó todos los: equipos de fabricación y control procesos, validación de limpieza, punto de apoyo crítico, métodos analíticos validados, instrumentos de laboratorio calificados y/o calibrados antes de la fabricación y del análisis de las muestras. Pese a lo anterior, se incluyó en el apartado de Metodologías de Validaciones la información acerca de cómo obtener los cuartiles para el análisis estadístico de los distintos lotes, que no estaban descritos anteriormente. Además, se modificó la toma de muestras en la etapa de lubricación, ya que previamente se tomaba una cantidad equivalente a 3,5 g de mezcla para cada uno de los puntos definidos en el mezclador. Al ser un producto de fabricación de compresión directa existe el riesgo de segregación del sólido, por ello se describe el procedimiento de tomar la cantidad equivalente a un 24.
(38) comprimido y que es trasvasijado a un matraz de aforo de 50 mL. Finalmente, el diagrama de flujo adjunto en el Protocolo de Validación permitió organizar de mejor manera todo el proceso de fabricación de Montelukast 5 mg comprimidos masticables. Aquello permite visualizar rápidamente cada una de las etapas del proceso junto con sus materias primas y equipos, frente a una revisión por parte de la entidad sanitaria. Por reserva de derecho de información con el laboratorio no se entregan más detalles de este, sin embargo, más adelante en la Figura N°4 se muestra un diagrama de flujo resumido del proceso de fabricación del producto en estudio. 3. Verificación del Proceso de Fabricación de Montelukast 5 mg Comprimidos Masticables Luego de actualizar los registros de fabricación provisorios para los 3 lotes y de confeccionar el protocolo de validación, con sus respectivos documentos adjuntos, se procedió con la verificación del proceso de fabricación de Montelukast 5 mg comprimidos masticables en el área de sólidos de la planta de producción, donde se acompañó y se brindó ayuda al operador a cargo en cada paso del proceso de manufactura. Además, se comprobó que se cumpliesen todos los pasos detallados en el registro de fabricación, PCC y se verificó que se ocuparan correctamente los equipos e instrumentos involucrados. 3.1 Metodología Para iniciar el proceso de fabricación, primero se comprobó que las materias primas mencionadas en la Tabla N°8 estuviesen disponibles y en la cantidad detallada en el registro de fabricación provisorio.. 25.
(39) Tabla N°8: Materias Primas utilizadas en las etapas de Pre-Lubricación y Lubricación. Etapa de Fabricación. Pre- Lubricación. Lubricación. Materias Primas. -. Montelukast Sódico. -. Sucralosa. -. Almidón glicolato. -. Saborizante de frutilla. -. Saborizante imitación chicle. -. Manitol. -. Manitol 200. -. Colorante óxido de hierro rojo. -. Ácido esteárico. -. Behenato de Glicerilo (Compritol 888). Además, se confirmó que los equipos correspondan a los citados en el registro de fabricación, estos debían tener las etiquetas respectivas de limpieza con fecha vigente. En la Tabla N°9 se mencionan de forma general los equipos e instrumentos utilizados durante el proceso productivo de Montelukast 5 mg comprimidos masticables. Tabla N°9: Equipos e Instrumentos utilizados durante el proceso de Fabricación de Montelukast 5 mg comprimidos masticables.. Etapa del Proceso de Fabricación. Fabricación granel Polvo. Equipo e Instrumentos -. Mezclador de 300 Litros. -. Mezclador de 20 Litros. -. Tamiz, mallas #30, #60 y #100. -. Balanza y espada de muestreo. 26.
(40) Continuación Tabla N°9… Etapa del Proceso de Fabricación. Compresión. Equipo e Instrumentos -. Tabletera Rotativa. -. Friabilómetro. -. Durómetro. -. Desintegrador. -. Balanza. 3.1.1 Toma de muestras en las etapas de Pre-lubricación y Lubricación Para realizar la toma de muestras se tuvo que saber el tamaño del mezclador que contenía a la mezcla de polvos a granel. Así se procedió a determinar el tamaño de la espada de muestreo a utilizar. Para un mezclador con una capacidad de 100 a 300 L se utiliza una espada pequeña de aproximadamente de 70 cm de largo. En contraste, se usó la espada grande de 110 cm aproximadamente para mezcladores de 800 a 1000 L de contenido. Para la fabricación de cada lote industrial de este producto se utilizó un mezclador de 300 L, por ende, se decidió utilizar la espada pequeña que se equipó con matrices de muestreo con un volumen de llenado de 3,5 mL y, matrices ciegas de forma intercalada a lo largo de la espada. Posteriormente, se procedió a tomar las muestras directamente desde el mezclador de acuerdo con la Figura 5. Donde se detalló la toma de 10 muestras tanto de la mezcla pre-lubricada como de la mezcla lubricada. Una vez finalizada la primera etapa de fabricación, se pesó una cantidad de 290,8 mg aproximadamente para cada muestra de la mezcla pre-lubricada, que estima el peso de un comprimido masticable de Montelukast 5 mg menos la masa del lubricante. Luego, al finalizar la lubricación se muestreó una cantidad de 300 mg equivalente a un comprimido. 27.
(41) Figura 4: Diagrama de Flujo del proceso de Fabricación de Montelukast 5 mg comprimidos masticables.. Manitol 200 Almidón glicolato Sabor frutilla Sabor imitación chicle. Tamizado 1 Tamiz Malla #30. Mezclado 1 Mezclador de 300 L. Separar mezcla en 2 partes. Parte 1. Manitol Col. Óxido de hierro rojo. Tamizado 2 Tamiz Malla #100 (2veces) y Malla# 30. Manitol Montelukast sódico Sucralosa. Tamizado 3 Tamiz Malla #60 y Malla# 30. Mezclado 2 Mezclador de 300 L Parte 2 Mezclado 3 Mezclador 300 L. Tamizado 4 Tamiz Malla #30. Compritol 888. Ácido Esteárico. Tamizado 5 y Premezcla Tamiz Malla #60 Mezclador de 20 L + 3,0 Kg de Mezcla Tamizado 6 y Premezcla Tamiz Malla #60 Mezclador de 20 L + 2,0 Kg de Mezcla. Lukanex 5 mg comp.. Mezclado 4 Mezclador de 300 L. Toma de muestra Prelubricación. Mezclado 5 Mezclador de 300 L. Mezclado 6 Final Mezclador de 300 L. Toma de muestra Lubricación. Compresión. Toma de muestra Compresión. 28.
(42) Cada una de las muestras fueron colocadas dentro de un matraz de aforo de 50 mL de vidrio ámbar. Se taparon y se rotularon correctamente para solicitar análisis de Homogeneidad de Montelukast en granel sin lubricar y lubricado. Se considera una especificación interna de 85%-115%, es decir, para cumplir con esta especificación cada muestra debía contener entre 4,25 – 5,75 mg de Montelukast.. Figura 5: Puntos de muestreo en mezclador de 300 L. Cada uno de los test detallados a continuación fueron ejecutados en las muestras de 50 g de mezcla recolectadas posterior a la lubricación, las cuales entregaron información acerca de las propiedades de flujo de la mezcla y ayudará a caracterizar el proceso de fabricación. 3.1.2 Ensayos reológicos 3.1.2.1 -. Densidad Densidad Aparente: Se determinó con una muestra de 30 g de. mezcla, luego se trasvasijó a una probeta de 100 mL y se midió el volumen ocupada por esta. -. Densidad a granel: La muestra ya contenida en la probeta de 100. mL se golpeó tres veces en una superficie sólida y firme desde una altura de 2,5 cm aproximadamente con un intervalo de 2 segundos por cada golpe y se midió el volumen final. 29.
(43) -. Densidad de Consolidación: Se determinó igual que la densidad. granel durante un minuto o hasta alcanzar un volumen constante.. Para calcular la densidad aparente y de consolidación se utilizó la siguiente fórmula: 𝐷=. 𝑀 𝑉𝐹. D: Densidad aparente o compactada de la mezcla (g/mL) M: Masa de la mezcla en gramos VF: Volumen final ocupado por el sólido en mL Luego de caracterizar la densidad aparente y compactada en etapa Lubricación se calcula el índice de compresibilidad y se comparó entre lotes mediante análisis de varianza ANOVA. Para aquello, se estimó los siguientes valores críticos de aceptación y de rechazo -. Si F (calculado) > F (de la tabla) significa que SI hay una diferencia. estadística significativa entre los lotes. Se rechaza la Hipótesis nula (H0). -. Si F (calculado) < F (de la tabla) significa que NO hay una diferencia. estadística significativa entre los lotes. Se acepta la Hipótesis nula. (H0). Donde: H0: No existe diferencia significativa de índice de compresibilidad entre los lotes fabricados bajo los parámetros establecidos en la etapa de Lubricación mezcla. 3.1.2.2. Granulometría. En primera instancia, se taró el conjunto de tamices ordenados de menor a mayor tamaño de partículas, luego se fue sacando de a uno y se registró la masa individual de cada tamiz. A continuación, se colocaron en el tamizador de forma ordenada y se incorporó una muestra de 50 g aproximadamente y 30.
(44) se sometió a 1 minuto de golpeteo y 5 minutos de vibración constante según el procedimiento operativo estándar propio del Laboratorio. Posteriormente, se pesaron nuevamente los tamices y por diferencia de masa se obtuvo la cantidad de muestra contenida en cada tamiz y se calcula el Área Superficial Específica Media (SSAM). Luego, se evaluó estadísticamente mediante un análisis de varianza ANOVA de dos factores con una sola muestra los datos de SSAM para distintas etapas y lotes. Se consideraron los mismos valores críticos de aceptación y rechazo descritos anteriormente en la densidad. Mientras que para la decisión estadística se estimó lo siguiente: -. Comparación entre etapas: F calculado es menor a F crítico. Se. acepta H0. -. Comparación entre lotes: F calculado es menor a F crítico. Se. Rechaza H0. Donde: Comparación entre etapas H0: No existe diferencia significativa en el valor de SSAM entre las etapas caracterizadas y fabricados bajo los parámetros establecidos.. Comparación entre Lotes H0: No existe diferencia significativa en el valor de SSAM entre los lotes fabricados bajo los parámetros establecidos. 3.1.2.3. Ángulo de reposo. Constituye una de las medidas más habituales para conocer la capacidad de flujo de un sólido. El flujo libre del sólido no solo depende de la fuerza gravitacional, sino que también de las fuerzas derivadas de la fricción interarticular, por lo que existe una estrecha relación entre el ángulo de 31.
(45) reposo, flujo y forma de la partícula. [11]. La correlación entre ángulo de reposo con la calidad de flujo se representó de acuerdo lo detallado en la Tabla N°10.. Tabla N°10: Propiedades de flujo de acuerdo con su ángulo de reposo Propiedades de Flujo. [11]. Ángulo de Reposo (°). Excelente. 25 – 30. Bueno. 31 – 35. Adecuado (no necesita ayuda). 36 – 40. Aceptable (puede demorarse). 41 – 45. Pobre (es necesario agitar o someter a. 46 – 55. vibración) Muy Pobre. 56 – 65. Extremadamente pobre. > 66. Para la medición del ángulo de reposo se requirió un embudo metálico ubicado a una altura conocida desde un soporte universal. Se utilizó una muestra de 30 g aproximadamente que se deja caer libremente por el embudo, una vez generado el cono de sólido se midió las extensiones de este, es decir, se mide la altura y el diámetro.. Mediante la siguiente fórmula se determinó el ángulo de reposo. 𝑡𝑎𝑛 𝛼 =. [11]. :. 𝐻 𝐷/2. Dónde tang α. :: Tangente del ángulo de reposo. H. :: Altura del cono. D :: Diámetro del cono DFFFFF Para verificar la reproducibilidad de proceso en esta etapa se realizó un 32.
(46) análisis de varianza ANOVA a los datos de ángulo de reposo obtenido entre los lotes de estudio. Para aquello se estimó las siguientes decisiones estadísticas: -. Si F (calculado) > F (de la tabla) significa que SÍ hay una diferencia. estadística significativa entre los lotes. Se rechaza la Hipótesis nula (H0). -. Si F (calculado) < F (de la tabla) significa que NO hay una diferencia. estadística significativa entre los lotes. Se acepta la Hipótesis nula. (H0). Dónde: H0: No existe diferencia significativa de ángulo de reposo entre los lotes fabricados bajo los parámetros establecidos en la etapa de Lubricación. 3.1.3 Toma de muestras en Etapa de Compresión De acuerdo con el tamaño de lote y velocidad de la tabletera, se calculó el tiempo total del proceso y luego este se dividió en 25 puntos para definir la frecuencia de muestreo, correspondiente a 60 comprimidos para cada punto. A cada muestreo se les realizó los siguientes ensayos en Planta Piloto de Investigación y Desarrollo: -. Peso (10 comprimidos). -. Dureza (10 comprimidos). El resto de los comprimidos se enviaron a Gestión muestra y se solicitan los siguientes ensayos al área analítica: -. Uniformidad de Montelukast (10 comprimidos). -. Valoración de Montelukast (20 comprimidos). -. Test de disolución (6 comprimidos). -. Descripción (10 comprimidos). De acuerdo con esto, se tomaron 25 puntos de 60 comprimidos cada 4 minutos para realizar los ensayos intralote e interlote de los ACC peso y 33.
(47) dureza en cada lote. No obstante, en el lote tres se tomó una cantidad de 24 puntos por falta de mezcla para el último punto. Cabe destacar que se tomaron tres puntos adicionales para cada lote que corresponden al inicio, medio y final de la compresión que se utilizaron para los ensayos de friabilidad y desintegración 3.1.4 Ensayos en Etapa de Compresión A las muestras obtenidas durante la compresión se les realizaron los siguientes test para verificar si es un proceso reproducible, controlado y si cumple con las especificaciones señaladas para cada Atributo Crítico de Calidad (ACC), mencionadas en la Tabla N°11. Para dar cumplimiento a las especificaciones de producto terminado se tuvo que tener en cuenta las características de la mezcla obtenida en etapas anteriores y por los parámetros de la tabletera. Estos últimos están sujetos a pequeñas intervenciones y/o ajustes durante el proceso de compresión, ya que se trabaja con cartas de control de peso, dureza y dimensiones del comprimido con un target específico para cada uno. Para obtener resultados acorte a los límites de establecidos para cada ACC se realizan controles periódicos cada 15 minutos y si llegase a ser necesario se modifican los parámetros de tabletera pero nunca superando los límites preestablecidos. Tabla N°11: Especificaciones de producto terminado registradas en ISP. Ensayos. Descripción. Peso. Rango Comprimidos de color rosado claro, levemente moteados, oblongos, biconvexos, ranurados en una cara y lisos por la otra Límite de especificación: 300 mg ± 5 % (285 – 315) mg Límite de control y validación: 300 mg + 3 % (291 – 309) mg. Procedimiento. Objetivo de la prueba. Inspección color, forma y presencia de ranura a 10 comprimidos de cada punto muestreado. Evaluación cumplimiento de ACC. Registrar el peso de 10 comprimidos en cada punto según frecuencia de muestreo. Evaluación cumplimiento de ACC y cumplimiento de análisis estadístico. 34.
(48) Continuación Tabla N°11… Ensayos. Rango. Procedimiento. Dureza. Límite de especificación: (3 – 30) Kp Límite de control: (6 – 13) Kp. Límite de Validación: (6-17) Kp. Registrar la dureza de 10 comprimidos en cada punto según frecuencia de muestreo. Realizar ensayo de desintegración a 6 comprimidos para cada lote, 3 determinaciones (Inicio, Medio y Final). Objetivo de la prueba Evaluación cumplimiento de ACC y cumplimiento de análisis estadístico Evaluación cumplimiento de ACC tanto para especificación como para control. Límite de especificación: < 30 min Límite de control y validación: < 15 min Límite de especificación: ≤ 1.0% Límite de control y validación: ≤ 0.5%. Realizar ensayo de desintegración a 6, 5 g de comprimidos para cada lote, 3 determinaciones (Inicio, Medio y Final). 90%-110% de lo declarado (4,5-5,5) mg/comp. Analizar 20 comprimidos (Gestión Muestra). Uniformidad de unidades de dosificación Montelukast. Valor de Aceptación ≤15. Medir uniformidad del activo obtenida durante la etapa de compresión (Gestión Muestra). Evaluación cumplimiento de ACC. Test de Disolución. No menos del 70% (Q) de la cantidad declarada se disuelve a los 45 minutos. Realizar el ensayo a 6 comprimidos de cada lote (Gestión Muestra). Evaluación cumplimiento de ACC. Desintegración. Friabilidad. Valoración de Montelukast. Evaluación cumplimiento de ACC tanto para especificación como para control Evaluación cumplimiento de ACC tanto para especificación como para control. 3.2 Resultados y Discusión 3.2.1 Ensayos en las etapas de Pre-lubricación y Lubricación Posterior a la toma de muestras en cada etapa, estas fueron enviadas a Gestión Muestra para solicitar el análisis de Homogeneidad de Montelukast para ambos graneles, los resultados obtenidos se detallan en la Tabla N°12. Se observó que para cada una de las muestras de los tres lotes verificados, 35.
Figure
![Figura 1: Variabilidad de un proceso por distintos factores involucrados [8] .](https://thumb-us.123doks.com/thumbv2/123dok_es/7321845.452257/19.918.175.780.565.846/figura-variabilidad-de-proceso-por-distintos-factores-involucrados.webp)
![Tabla N°1: Valores de Cp y su interpretación [8] .](https://thumb-us.123doks.com/thumbv2/123dok_es/7321845.452257/22.918.180.771.225.524/tabla-n-valores-cp-interpretación.webp)
![Figura 2: Elementos de una carta de control [8] . 4.7. Histograma](https://thumb-us.123doks.com/thumbv2/123dok_es/7321845.452257/25.918.180.770.363.700/figura-elementos-de-una-carta-de-control-histograma.webp)
![Figura 3: Estructura de Montelukast sódico (MTK) [15]](https://thumb-us.123doks.com/thumbv2/123dok_es/7321845.452257/28.918.184.768.193.434/figura-estructura-montelukast-sódico-mtk.webp)
+7




Documento similar
Perros: Indicado para el tratamiento de infecciones de la piel y tejidos blandos (pioderma de los pliegues cutáneos, impétigo, foliculitis, forunculosis, celulitis) causadas por
.Evaluando las tres etapas se obtuvo un Cp de 1.87 y Cpk de 1.87, confirmando con estos resultados que el proceso de fabrication de Brimodin 100 mg / 5 mL Jarabe e s
El marbofloxacino es efectivo frente una amplia gama de bacterias Gram positivas (concretamente frente a Staphylococcus spp., Streptococci spp.) y Gram negativas
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú.. Para ver una copia de dicha licencia,
Esta obra ha sido publicada bajo la licencia Creative Commons Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/... Biblioteca Digital
MEZCLA FINAL - 43 Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú.. Para ver una copia de
Para el parámetro de peso promedio, los resultados obtenidos de los 3 lotes cumplen con la especificación establecida lo que demuestra que el proceso se encuentra bajo control y 25
En el estudio de capacidad de proceso Cp para viscosidad se dio durante la producción de los tres lotes, esto se dio durante el proceso de fabricación en el análisis de la muestra
